The presentdisclosurerelatestoanadhesivecomposition
and amethodforpreparingthesame,andmoreparticularly,
to anadhesivecompositionhavinghighbiodegradability
and excellentmechanicalpropertieswhilebeingbiocompat-
ible, andamethodforpreparingthesame
Incorporating rubbery domains into glassy polymers is an effective route to improve toughness and impact strength. However, retaining the transparency of the composite material over a wide temperature range with enhanced mechanical attributes is challenging because of a mismatch in refractive indices with changing temperatures, limiting their applications as optical materials. Here, we report photopolymerization-induced microphase separation as a strategy for the fabrication of transparent, temperature-resistant nanostructured polymeric materials. Taking poly(methyl methacrylate) (PMMA) as the glassy component for its renowned high transparency, we perform controlled radical polymerization upon light exposure to transform the whole polymerization mixture into a crosslinked block polymer material, where a bicontinuous nanostructure consisting of PMMA and cross-linked rubbery
microdomains spontaneously arises during polymerization in situ. The facile formation of the rubbery domains, smaller than 50 nm yet 3D continuous and cross-linked, is the key to retaining transparency above 120 °C in the visible light wavelength with dimensional stability and allowing efficient stress dissipation through the large interfacial area. We further demonstrate the 3D printability of the nanostructured materials into custom shapes via direct ink writing.
Nature utilizes self-assembly to form complex, functional structures, inspiring advanced materials design. Polymer crystallization drives assemblies with both ordered and disordered regions. Crystallization-driven assembly of BCPs enables unique hierarchical nanostructures with enhanced colloidal stability and directionality, applicable from optoelectronics to biomedicine. However, mechanisms governing morphological transitions remain poorly understood due to complex microphase separation and competitive crystallization. Using liquid-phase transmission electron microscopy, we visualize the spontaneous assembly of semicrystalline amphiphilic BCPs. We observe structural transformations from unimers to spherical, cylindrical, toroidal micelles, and vesicles by varying constituent block ratios. Image segmentation overcomes low contrast of aqueous assemblies, enabling motion tracking. Nanostructures exhibit structural evolution driven by long-range hydrophobic interactions from formed elemental micelles undergoing anomalous diffusion. Notably, toroid formation follows a distinct pathway compared with conventional BCPs due to semicrystalline BCPs’ preference for low curvature at the core-corona interface. Insights into assembly dynamics via real-time imaging provide strategies for controlling complex hierarchical structures.
The C–H functionalization of polymers enables the direct incorporation of new functional groups into polymer backbones, presenting significant opportunities for the upcycling of commodity polymers. However, developing reactions that achieve selective functionalization while preserving the intrinsic features of polymers and avoiding undesirable structure deformation remains a considerable challenge. In this study, we present a transition metal-free post-functionalization approach for polyethers via a photoinduced α-C–H amidation reaction. This strategy provides a route to previously unattainable α-amino polyethers, which exhibit distinct physical properties from those of the parent polymer. By leveraging a polar-radical relay mechanism, we effectively incorporate C–N bonds into the polyether backbone while suppressing degradation and cross-linking. Conducted under mild and convenient conditions, this approach demonstrates significant site selectivity at the ethereal α-position, even in the presence of other types of C–H bonds, achieving tailed post-functionalization of macromolecules. Furthermore, the present strategy holds promise for broader applications, including the amidative degradation of commodity polymers and transformation of polyethylene glycol (PEG) network.
Permselective anion exchange membranes (AEMs) are of great interest in electrochemical devices, allowing the selective transport of species of interest through the membrane while preventing the crossover of undesired substrates. However, porous and nonporous membranes have traditionally been developed and applied separately for these applications. In this study, we utilized the merits of polymerization-induced microphase separation to observe the transition from porous to nonporous membranes by controlling the etchable block length and the hydrophilic domain size, respectively. At a certain point, a transition from porous to nonporous permeation behavior was observed, where permeation no longer depended on the open pore size. For the fabrication of AEMs with well-defined and 3D continuous nanochannels, we integrated an etchable sacrificial block with a pre-functional block containing precursors for quaternary ammonium groups to create a pore decorated with positively charged dangling chains. Etching and amination yielded the target AEM with a controlled pore size and ammonium ion density. Increasing the length of the dangling chains resulted in nonporous AEMs, which allowed exclusive permeation of a negatively charged dye over a positive one. This provides strong evidence that the solution-diffusion model applies to nonporous membranes. Tunable permselectivity over a wide range, combined with stability in organic solvents and alkaline conditions, suggests that this methodology holds significant potential for the development of membranes for advanced electrochemical systems.
Despite extensive studies on nanoporous membranes for regulating lithium-ion (Li⁺) flux in lithium (Li)-metal batteries, the pore size design has largely focused on very small (< 5 nm) or extremely large (> 20 nm) dimensions, overlooking the intermediate pore size range. This gap, particularly between 5 and 15 nm, has limited exploration of critical Li⁺ transport phenomena and their impact on improving cell performance. Here, we developed robust and free-standing polymeric films with three-dimensional (3D) continuous nanoporous channels, precisely tuned to pore diameters ranging from 5 to 14 nm and immobilized sulfonate groups. Our systematic investigations revealed how pore size and immobilized anionic groups correlated with Li⁺ conductivity and battery performance. Notably, sulfonate-functionalized channels promoted Li⁺ conductivity significantly within this optimal pore range compared to non-functionalized counterparts. In an ether-based electrolyte with 1 M lithium bis(fluorosulfonyl)imide (LiFSI), the Li⁺ conductivity peaked at a pore diameter of 10 nm. Furthermore, the mobility of Li⁺ was approximately 4.4 times faster than FSI⁻, resulting in reducing interfacial resistance and promoting uniform Li deposition. The sulfonated nanoporous membrane in Li|LiFePO₄ full cells with an N/P ratio of 2.3 delivered excellent cycling stability over 1000 cycles while retaining approximately 80 % of the initial capacity.
Multicompartment polymer nanoparticles, such as two-faced Janus and patchy particles composed of distinct chemical features, have received increasing attention because of their utility for interfacial and self-assembly applications originating from the asymmetric particulate structure. This review discusses such nanoparticles in several tens of nanometers produced by controlled polymerizations, which enable scalable synthesis with control of molecular characteristics. We focus on miktoarm core cross-linked star polymers and Janus core–shell bottlebrush polymers as 0D and 1D anisotropic nano-objects containing a discrete core and a compartmentalized shell. We discuss how controlled polymerizations can covalently build such complex architectures with spatial control of the constituting segments to achieve intramolecular segregation. Then, we collectively view their distinct interfacial and self-assembling behaviors reported in the literature from experimental and simulation perspectives.
Polymer grafting density critically influences the self-assembly of polymer-grafted nanoparticles, yet the low grafting density regime remains underexplored. Here, we investigate the thin-film self-assembly of bottlebrush polymer-grafted core/shell nanoparticles (BPGNPs) under quasi-2D confinement at near-zero grafting densities through coarse-grained molecular dynamics (CGMD). The NP core is modeled using a hard-core/soft-shoulder (HCSS) potential, and it is compared against Weeks–Chandler–Andersen (WCA) potential. While the phase behaviors of both models are well-known, the distinct phase behaviors of both models persist even with polymer grafting offering additional room for tunability. Unlike sufficiently high grafting density or bare nanoparticles (NPs), grafting a single bottlebrush polymer breaks the rotational symmetry. The resulting structural polarity of grafted NPs can be precisely controlled through bottlebrush design parameters. We demonstrate that enhanced structural polarity stabilizes specific ordered phases, enabling precise control over self-assembled morphologies such as hexagonal lattices, square lattices, and linear clusters. Lastly, we explore the impact of synthesis-induced heterogeneity by introducing bare NPs, dual-polymer-grafted particles, and unconjugated polymers as minor species, providing insights into morphological stability under realistic grafting conditions. This work advances our understanding of BPGNP self-assembly in the near-zero grafting density regime and establishes design principles for functional nanotechnology applications.
We report a new bottlebrush copolymer (BBCP) ligand design as robust patches for gold nanoparticles (Au NPs) to construct a rigid template guiding heterometal deposition on the surface. Given the spatial congestion of the side chains, the BBCP rapidly forms dense and stationary patches on Au NPs and effectively blocks additional metal deposition. Reducing solvent quality varies the phase segregation of the BBCP and subsequently restricts metal deposition to specific locations, fabricating diverse bimetallic heterostructures. The resulting morphology exhibits a unique orientation-dependent scattering property that thermodynamic configuration cannot achieve.
Accumulation of aquatic organisms on submerged sea vessel surfaces increases drag, resulting in higher fuel consumption and more greenhouse gas emissions. Polymer coatings hold promise to combat marine biofouling as more environmentally friendly alternatives to metal-based paints. While hydrophilic coatings are known to lose antifouling efficacy upon sediment adsorption, how to arrange hydrophobic segments and develop a sufficiently thick coating layer to suppress diatom deposition in the presence of silt and maximize the antifouling performance has been unknown. Here, we developed an effective and scalable antifouling coating by combining the polydopamine precoating method with well-defined amphiphilic copolymers synthesized using a controlled polymerization technique. Reversible addition-fragmentation chain transfer copolymerization of zwitterion-containing hydrophilic sulfobetaine methacrylate and hydrophobic trifluoroethyl methacrylate produced the target copolymers with control of composition, molecular weight, and sequence. Zr(IV)-mediated coordination bonds between polydopamine and sulfobetaine groups yielded >20 nm thick films stable for a month in seawater. The random copolymer sequence exposed both hydrophilic and hydrophobic groups on the outermost coating surface. Synergistic repelling diatom and silt led to the best antifouling performance at the optimal hydrophilic-hydrophobic balance. Superior antifouling efficacy was retained for a month and was also effective on stainless steel, suggesting the potential for practical application.
We report growing a polymer chain from the backbone of a bottlebrush polymer in the neat polymerization condition produces nanostructured polymer monoliths with ordered morphologies based on the Janus bottlebrush architecture. We installed a norbornene unit at the end of the polylactide macro-chain transfer agent (PLA-CTA) by single unit monomer insertion. We polymerized the resulting macromonomer via ring-opening metathesis polymerization to produce the PLA bottlebrush polymer, where a trithiocarbonate moiety remains on the backbone per every repeating unit. Neat polymerization of styrene in the presence of the PLA bottlebrush polymer proceeded in a grafting-from manner following the reversible addition–fragmentation chain transfer mechanism, resulting in a monolithic solid containing the doubly grafted PLA and polystyrene (PS) side chains. Polymerization-induced microphase separation (PIMS) spontaneously occurred, driven by the incompatibility between PLA and the growing PS segment. In contrast to the significant disordered fraction in PLA-b-PS produced with the linear PLA-CTA, the PLA/PS Janus bottlebrush polymer showed improved order across the investigated composition range. Formation of the asymmetric lamellae up to >80 vol % of PS indicated a strong preference for the lamellar symmetry of the Janus architecture. The in situ structured monoliths even exhibited narrower scattering peak widths compared to the solution-cast and annealed sample, suggesting the utility of the Janus PIMS process for facile preparation of ordered nanostructured materials with uniform domain size.
Macromolecular self-assembly is essential in life and interfacial science. A macromolecule consisting of chemically distinct components tends to self-assemble in a selective solvent to minimize the exposure of the solvophobic segments to the medium while the solvophilic segments adopt extended conformations. While micelles composed of linear block copolymers represent classic examples of such solution assembly, recent interest focuses on the self-assembly of complex macromolecules with nonlinear architectures, such as star, graft, and bottlebrush. Such macromolecules include several to hundreds of polymer chains covalently tied to a core and a backbone. The pre-programmed, non-exchangeable chain arrangement makes a huge difference in their self-assembly. The field has witnessed tremendous advances in synthetic methodologies to construct the desired architectures, leading to discoveries of exotic self-assembly behavior. Thanks to the rapid evolution of computing power, computer simulation has also been an emerging and complementary approach for understanding the association mechanism and further predicting the self-assembling morphologies. However, simulating the self-assembly of architected macromolecules has posed a challenge as a huge number of objects should be included in the simulations. Comparing experimental results with simulations is not always straightforward, as synthetic routes to well-defined model systems with systematically controlled structural parameters are not often available. In this manuscript, we propose to bridge a gap between experiments and simulations in self-assembly of architected macromolecules. We focus on the key articles in this area reporting experimental evidence and simulation details and also cover recent examples in the literature. We start with discussing simulation methodologies applicable to investigate solution self-assembly across multiple levels of chemical resolution from all-atom to particle dynamics. Then, we delve into topological design, synthesis, and simulation of nonlinear macromolecules, including dendritic/star, network, and graft/bottlebrush polymers, to understand the architectural effect on the self-assembly behavior. We expand our discourse to embrace recent advances toward realizing more complex systems. For example, self-assembly in the presence of strong Coulombic interactions, such as in the case of polyelectrolytes, geometric constraints, and other components in solutions, exemplified by inorganic fillers, are introduced. Finally, the challenges and perspectives are discussed in the final section of the manuscript.
We investigated the bilayer-folded lamellar (Lf) mesophase appearing in the aqueous solution of amphiphilic random copolymers. A series of copolymers were synthesized by reversible addition–fragmentation chain transfer copolymerization of oligo(ethylene glycol) acrylate with alkyl acrylate with different alkyl chain lengths from octyl (C8) to octadecyl (C18). The alkyl acrylate composition was adjusted between 50–60 mol %. In the concentrated solution with the carbon number of the alkyl side chain higher than 10, the copolymers associated in water via hydrophobic interaction between the alkyl chains to produce micellar bilayers, which were periodically folded into bilayer-folded lamellae. The appearance of a small-angle X-ray scattering (SAXS) peak at a low scattering vector corresponding to >10 nm length scale clearly distinguished the bilayer-folded lamellae from the micellar lamellae with the domain spacing of 5–7 nm. Two-dimensional (2D) SAXS corroborated the presence of bilayer-folded lamellae developing perpendicularly to the micellar lamellae, which is consistent with our previous report. While the Lf phase was observed at room temperature for dodecyl (C12) and tetradecyl (C14) side chains that formed amorphous packing, crystalline hexadecyl (C16) and octadecyl (C18) chains seem to disturb bilayer folding. Heating the solution above the melting temperature of the alkyl chains produced the Lf phase with the largest folding height in the case of C16. The scaling relationship of the folding height to the carbon number supports the idea that the bending rigidity of the bilayer influences the length scale of folding.
In this study, we present extensive dissipative particle dynamics simulation studies of bottlebrush copolymers in solution having different grafting sequences: block and random. Distinct morphology of the grafting sequence-controlled bottlebrush copolymer micelles is investigated through backbone chain distribution along with the micelle structure. As a result, bottlebrush block copolymer (BBCP) micelles exhibit backbone chain primarily dependent on length scale of micelle size, while bottlebrush random copolymer (BRCP) exhibits side chain-dependent length scale. We further quantify the dependence of the micelles on the length scale of the backbone chain and side chain using the scaling relationship. We decouple the size of the micelles into core radius and corona thickness, and scaling behavior of these structures is quantitatively explained by the conformation of backbone chains and side chains. Also, the experimental scaling of BBCP and BRCP micelles in water shows consistent results of the sequence-dependent scaling exponents calculated by simulation. This work reveals the scaling behavior of the sequence-controlled graft copolymer micelles which potentially guides how one can modify the solution self-assembled complex micelles by controlling architecture and structure parameters of the bottlebrush copolymer.
We report the synthesis of the amphiphilic diblock copolymer, poly (vinyl phenol)-block-poly (vinyl alcohol), and its reversible formation of invertible core-shell nanoparticles. The diblock copolymer composed of poly (4-vinylphenol) (P4VPh) and poly (vinyl alcohol) (PVA) was prepared by the switchable reversible addition-fragmentation chain transfer polymerization (switchable-RAFT) of 4-acetoxystyrene and vinyl acetate and subsequent acetyl deprotection. The diblock copolymer was soluble in dimethyl sulfoxide (DMSO) but self-assembled into core-shell nanoparticles with the addition of H2O or tetrahydrofuran (THF). The formation and transition of the invertible core-shell system in response to solvent composition was characterized by 1H NMR, dynamic light scattering, and transmission electron microscopy. Interestingly, the fluorescent behavior of the P4VPh block was changed according to the morphological change of core-shell nanoparticle induced by solvent composition. With increasing the H2O content, the P4VPh chains in the DMSO solution collapsed and the diblock copolymers self-assembled to core-shell type micelles. Aggregation of P4VPh chains increased the local concentration of the phenolic group, resulting in fluorescence quenching. However, with the addition of THF to the DMSO solution, the inverted core-shell nanoparticles were formed where segregated P4VPh chains showed fluorescence.
Depolymerization breaks down polymer chains into monomers like unthreading beads, attracting more attention from a sustainability standpoint. When polymerization reaches equilibrium, polymerization and depolymerization can reversibly proceed by decreasing and increasing the temperature. Here, we demonstrate that such dynamic control of a growing polymer chain in a selective solvent can spontaneously modulate the self-assembly of block copolymer micellar nano-objects. Compared to polymerization-induced self-assembly (PISA), where irreversible growth of a solvophobic polymer block from the end of a solvophilic polymer causes micellization, polymerization/depolymerization-induced self-assembly presented in this study allows us to reversibly regulate the packing parameter of the forming block copolymer and thus induce reversible morphological transitions of the nano-objects by temperature swing. Under the coupled equilibria of polymerization with self-assembly, we found that demixing of the growing polymer block in a more selective solvent entropically facilitates depolymerization at a substantially lower temperature. Taking ring-opening polymerization of δ-valerolactone initiated from the hydroxyl-terminated poly(ethylene oxide) as a model system, we show that polymerization/depolymerization/repolymerization leads to reversible morphological transitions, such as rod–sphere–rod and fiber–rod–fiber, during the heating and cooling cycle and accompanied by changes in macroscopic properties such as viscosity, suggesting their potential as dynamic soft materials.
Thermo-responsive diblock copolymer, poly(N-isopropylacrylamide)-block-poly(N-vinylisobutyramide) was synthesized via switchable reversible addition–fragmentation chain transfer (RAFT) polymerization and its thermal transition behavior was studied. Poly(N-vinylisobutyramide) (PNVIBA), a structural isomer of poly(N-isopropylacrylamide) (PNIPAM) shows a thermo-response character but with a higher lower critical solution temperature (LCST) than PNIPAM. The chain extension of the PNVIBA block from the PNIPAM block proceeded in a controlled manner with a switchable chain transfer reagent, methyl 2-[methyl(4-pyridinyl)carbamothioylthio]propionate. In an aqueous solution, the diblock copolymer shows a thermo-responsive behavior but with a single LCST close to the LCST of PNVIBA, indicating that the interaction between the PNIPAM segment and the PNVIBA segment leads to cooperative aggregation during the self-assembly induced phase separation of the diblock copolymer in solution. Above the LCST of the PNIPAM block, the polymer chains begin to collapse, forming small aggregates, but further aggregation stumbled due to the PNVIBA segment of the diblock copolymer. However, as the temperature approached the LCST of the PNVIBA block, larger aggregates composed of clusters of small aggregates formed, resulting in an opaque solution.
Kim, Namhee; Kang, Jun Su; Jun, Taesuk; Suh, Jong-Min; Roh, Deok-Ho; Park, Won-Woo; Kwon, Oh-Hoon; Kwon, Tae-Hyuk; Lim, Mi Hee; Ryu, Du Yeol; Seo, Myungeun; Kim, Byeong-Su
This study develops a new type of C3-symmetric triphenylene triimide (TTI) bearing different oligo(ethylene glycol) side chains via imide linkages. By exploiting the unique TTI molecule as a building block, supramolecular polymerization is explored based on π–π stacking and hydrophilic/hydrophobic interactions in various solvents and the rates of heating/cooling process. The molecular chirality of the TTI unimer induces a preferential helicity formation in fibrous structures, while the achiral side chain allows the formation of linear nanofibers. The stacking type of supramolecular polymerization is highly dependent on the point chirality of the side chains, as indicated by the spectroscopic analyses, including ultraviolet–visible (UV/vis) and circular dichroism (CD) spectroscopy with atomic force microscopy (AFM), transmission electron microscopy (TEM), and wide-angle X-ray scattering (WAXS). Interestingly, the supramolecular polymerization does not occur in its monomeric state due to the generation of radical anions from the imide groups upon UV irradiation. In contrast, the fibrous structure in the assembled state is maintained, owing to the intermolecular interaction. This study provides a new direction in the phototriggered control of the supramolecular chiral assembly.
Block polymers comprising covalently joined polymeric segments represent a class of nanostructured, multicomponent polymeric materials. Polymerization-induced microphase separation (PIMS) is an intriguing subset that allows for simultaneous nanostructuring during block polymer synthesis. In contrast to polymerization-induced self-assembly (PISA), useful for the spontaneous formation of block polymer micelles, PIMS is well suited to fabricating monolithic block polymer materials by turning a whole polymerization mixture into a nanostructured solid. With the in situ cross-linking feature, PIMS offers a facile route to nanostructured block polymer thermosets in combination with various polymerization techniques, from emulsion polymerization to 3D printing. This review aims to provide a comprehensive overview and practical guide on PIMS by covering its historical background and mechanistic aspects and also highlighting representative material classes and applicable polymerization techniques.
개시된 3차원 계층적 나노구조체의 제조 방법은, 다공성 주형을 형성하는 단계, 상기 다공성 주형의 기공 내에 블록공중합 용액을 제공하는 단계, 상기 블록공중합 용액의 중합 반응을 진행하여, 상기 다공성 주형의 역상을 형상을 가지며 서로 다른 용해 특성을 갖는 지지블록과 희생블록을 포함하는 상분리된 3차원 구조체를 형성하는 단계, 상기 다공성 주형을 제거하는 단계 및 상기 상분리된 3차원 구조체의 희생블록을 제거하는 단계를 포함하는 3차원 계층적 나노구조체의 제조방법을 포함한다. 이에 따르면, 다양한 응용분야에서 필요로 하는 최적화된 형태와 크기의 나노구조를 제공할 수 있다.
The present invention relates to a method of synthesizing hydrocarbon polymers using a deoxygenation reaction, wherein, by deoxygenating polymers including oxygen atom-containing functional groups in side chains thereof to thereby remove the functional groups of the side chains, various block copolymers including polyolefins and hydrocarbon polymers with complex architectures can be synthesized.
We developed a synthetic route, based on radical polymerization, to a fluorescent monolithic hierarchically porous polymer composed of extended π-conjugated triphenylene motifs. A hexa-vinyl cross-linker containing the triphenylene core was synthesized and copolymerized with styrene in the presence of a polylactide macro-chain transfer agent to produce a cross-linked block copolymer monolith. Polymerization-induced microphase separation occurred during polymerization in situ, resulting in a disordered bicontinuous morphology of polylactide and cross-linked polystyrenic domains at a nanometer scale. Removal of polylactide generated percolating mesopores with controllable pore size and exposed micropores within the polystyrenic network. A strong bluish fluorescence was observed from the resulting porous monolith, originating from the embedded triphenylene. Fluorescence was quenched upon exposure to a solution of nitroaromatic compounds. Much stronger and faster quenching compared to the nonporous analog was attributed to the improvement in access to the triphenylene group via enhanced diffusion of the analyte through the interconnected mesopores.
본 발명은 탈산소화반응을 이용한 탄화수소계 고분자의 합성방법에 관한 것으로서, 산소원자를 지니는 관능기를 측쇄에 포함하는 고분자의 탈산소화 반응을 이용해 측쇄의 관능기를 제거하여 폴리올레핀을 포함하는 다양한 블록 공중합체 및 복잡한 아키텍처의 탄화수소계 고분자를 합성할 수 있다.
Bioadhesives are becoming an essential and important ingredient in medical science. Despite numerous reports, developing adhesive materials that combine strong adhesion, biocompatibility, and biodegradation remains a challenging task. Here, we present a biocompatible yet biodegradable block copolymer-based waterborne superglue that leads to an application of follicle-free hair transplantation. Our design strategy bridges self-assembled, temperature-sensitive block copolymer nanostructures with tannic acid as a sticky and biodegradable polyphenolic compound. The formulation further uniquely offers step-by-step increases in adhesion strength via heating–cooling cycles. Combining the modular design with the thermal treating process enhances the mechanical properties up to 5 orders of magnitude compared to the homopolymer formulation. This study opens a new direction in bioadhesive formulation strategies utilizing block copolymer nanotechnology for systematic and synergistic control of the material’s properties.
Micrometer-sized aqueous droplets serve as a unique reactor that drives various chemical reactions not seen in bulk solutions. However, their utilization has been limited to the synthesis of low molecular weight products at low reactant concentrations (nM to μM). Moreover, the nature of chemical reactions occurring outside the droplet remains unknown. This study demonstrated that oil-confined aqueous microdroplets continuously generated hydroxyl radicals near the interface and enabled the synthesis of polymers at high reactant concentrations (mM to M), thus successfully converting the interfacial energy into the synthesis of polymeric materials. The polymerized products maintained the properties of controlled radical polymerization, and a triblock copolymer with tapered interfaces was prepared by the sequential addition of different monomers into the aqueous microdroplets. Furthermore, a polymerization reaction in the continuous oil phase was effectively achieved by the transport of the hydroxyl radicals through the oil/water interface. This interfacial phenomenon is also successfully applied to the chain extension of a hydrophilic polymer with an oil-soluble monomer across the microdroplet interface. Our comprehensive study of radical polymerization using compartmentalization in microdroplets is expected to have important implications for the emerging field of microdroplet chemistry and polymerization in cellular biochemistry without any invasive chemical initiators.
We report a synthetic methodology for decorating a surface of metal–organic frameworks (MOFs) with polymers through postsynthetic modification. Well-defined polymers with reversibly deactivated radical species at their chain end were reacted with vinyl-functionalized MOFs in the presence of a radical initiator. The radical addition forms a C–C bond between the polymer end with the functional group at the MOF ligand. We used sterically bulky star polymers containing electron-deficient maleimide chain ends, which facilitated modification of the external surface, yielding polymer-grafted MOF composite particles. A patchy MOF particle can also be obtained by simultaneously grafting two polymers and jammed at the immiscible liquid–liquid interface. We further show that the selective removal of a sacrificial polymer would partially expose the surface of MOFs to external environment, which hinders the uptake of macromolecular guests above the critical hydrodynamic size. Overall, four polymer@MOF composites have successfully been achieved through the present postsynthetic patchworks on MOFs with star polymers and selective etching process.
Randomness is perceived in two different extremes, in macroscopic homogeneity and local heterogeneity, but apparently far away from order. Here we show that a periodic order spontaneously arises from a binary random copolymer when self-assembly occurs in an ensemble containing > 1015 possible chain sequences. A Bernoullian distribution of hydrophilic and hydrophobic side chains grafted onto a linear backbone was constructed by random copolymerization. When the polymer chains associate in water, a sequence matching problem occurs because of the drastic heterogeneity in sequence: this is believed to generate local curvature mismatches which deviate from the ensemble-averaged interfacial curvature. Periodic folding of the self-assembled bilayer stabilizes the curvature instability as recurring hinges. Reminiscent of chain-folded lamellae found in polymer crystallization, this new liquid crystalline mesophase, characterized as bilayer-folded lamellae, manifests itself as an anisotropically alignable birefringent hydrogel with structural hierarchy across multiple length scales.
We report the synthesis and self-assembly of brush–linear diblock copolymers with variable side-chain length and density. Poly(pentafluorophenyl acrylate-g-ethylene glycol)-b-polystyrene ((PPFPA-g-PEG)-b-PS) brush–linear diblock copolymers are prepared by sequential reversible addition–fragmentation chain transfer (RAFT) polymerization of PPFPA and PS, followed by postpolymerization reaction between the precursor PPFPA-b-PS diblock copolymer and amine-functionalized PEG. By controlling the PEG chain length and the degree of substitution, we obtained brush–linear diblock copolymers with different side-chain lengths and densities. The solid-state morphologies of the diblocks are then examined by small-angle X-ray scattering (SAXS). At low PEG side-chain density, the segregation of PEG and PS away from PPFPA leads to the formation of PEG and PS lamellar domains with PPFPA in the interface. At high PEG side-chain density, the segregation is between the PPFPA-g-PEG brush block and the PS linear block, and the domain morphology is determined by the composition of the brush block. A partial experimental phase diagram is presented, and it illustrates the importance of both side-chain length and density on the microdomain morphology of brush–linear diblock copolymers.
Bottlebrush polymers (BBPs) are a type of comb-like macromolecule with densely grafted polymeric sidechains attached to the polymer backbones, and many intriguing properties and applications have been demonstrated due to their unique architecture. Moreover, a ring-opening metathesis polymerization (ROMP) technique using Grubbs catalysts allows a precise control of various structural parameters in BBPs, such as the sidechain length, backbone length, and sidechain microstructures. This review mainly highlights recent advances of BBPs prepared by ROMP, from synthesis efforts to properties and applications.
Circularly polarized light (CPL) is an inherently chiral entity and is considered one of the possible deterministic signals that led to the evolution of homochirality. While accumulating examples indicate that chirality beyond the molecular level can be induced by CPL, not much is yet known about circumstances where the spin angular momentum of light competes with existing molecular chiral information during the chirality induction and amplification processes. Here we present a light-triggered supramolecular polymerization system where chiral information can both be transmitted and nonlinearly amplified in a “sergeants-and-soldiers” manner. While matching handedness with CPL resulted in further amplification, we determined that opposite handedness could override molecular information at the supramolecular level when the enantiomeric excess was low. The presence of a critical chiral bias suggests a bifurcation point in the homochirality evolution under random external chiral perturbation. Our results also highlight opportunities for the orthogonal control of supramolecular chirality decoupled from molecular chirality preexisting in the system.
Supramolecular polymerization offers a fascinating opportunity to develop dynamic soft materials by associating monomeric building blocks via noncovalent interactions. We report that polymerization can spontaneously drive the supramolecular polymerization of nanoscale micellar objects. We constructed the patchy micelles via two-step polymerization-induced self-assembly. A horizontal association between the patches results in a 1D supermicellar chain in situ by minimizing the enthalpic penalty of exposing the growing chains to solvent. Its length grows with increasing degree of polymerization, confirming that the supramolecular polymerization was triggered and controlled by polymerization. Our results highlight the observation that (1) the entire self-assembly process of forming, compartmentalizing, and associating the micelles can be driven by polymerization in a concerted manner and that (2) polymerization-induced self-assembly now can use compartmentalized nanoobjects as substrates beyond block copolymer chains. Polymerization-induced supramolecular polymerization could be useful for the autonomous preparation of hierarchical nanostructures.
Stereoselective synthesis of polylactic acid (PLA) was achieved using titanium(IV) complexes. The NO3 ligands prepared from 2,2′-dihydroxybenzophenone and salicylaldehyde derivatives were used to control the stereoselectivity of Ti(IV)-catalyzed polymerization of rac-lactide, in which a substituted ligand provided heterotactic PLA with high stereoselectivity. Density functional theory calculations revealed that the ligand structure is crucial for differentiating reaction pathways, and that the octahedral transition states are stabilized by the preorganized intermediates.
Introduction of asymmetry into a supramolecular system via external chiral stimuli can contribute to the understanding of the intriguing homochirality found in nature. Circularly polarized light (CPL) is regarded as a chiral physical force with right- or left-handedness. It can induce and modulate supramolecular chirality due to preferential interaction with one enantiomer. Herein, this review focuses on the photon-to-matter chirality transfer mechanisms at the supramolecular level. Thus, asymmetric photochemical reactions are reviewed, and the creation of a chiral bias upon CPL irradiation is discussed. Furthermore, the possible mechanisms for the amplification and propagation of the bias into the supramolecular level are outlined based on the nature of the photochromic building block. Representative examples, including azobenzene derivatives, polydiacetylene, bicyclic ketone, polyfluorenes, Cn-symmetric molecules, and inorganic nanomaterials, are presented.
The present invention relates to a method of preparing a hierarchically porous polymer and a hierarchically porous polymer prepared thereby. The method comprises the steps of: (a) polymerizing an external oil phase of a high internal phase emulsion (HIPE) consisting aqueous droplets to produce a cross-linked block copolymer; (b) obtaining a macroporous polymer with interconnected macropores by removing the aqueous droplets; and (c) treating the obtained porous polymer with a base, thereby obtaining a hierarchically porous polymer having three-dimensional mesopores formed in the macroporous walls. According to the method, the macropore size and mesopore size of the hierarchically porous polymer can all be controlled. The hierarchically porous polymer prepared by the method can easily separate polymers having different sizes, and thus is highly useful in the polymer separation field.
The glass-transition temperature (Tg) and degree of crystallinity (Xc) are important properties of polymers. However, for some polymers, current techniques have some difficulties in measuring Tg and Xc, particularly for as-prepared films without damage. Here, we develop a new technique to simultaneously determine Tg and Xc by measuring the restitution of a ball on an intact polymer film or sheet as a function of temperature. Demonstrating with eight different types of polymers, we show that Tg is the onset of the decrease in restitution, and Xc is the minimum restitution for semi-crystalline polymers with Xc up to 50%. Our simple yet versatile technique could provide a useful tool to measure challenging Tg and Xc of films and sheets by the conventional methods.
We developed a methodology, inspired by the folding of proteins, for the precision synthesis of hairy polymer nanoparticles. High-molar mass and narrowly dispersed graft copolymers were synthesized by graft-through ring opening metathesis polymerization, to incorporate a designated number of side chains and dimerizable cinnamic acid groups. Intrachain photodimerization collapsed the backbone and arrested it into a compact globular conformation, resulting in hairy nanoparticles topologically equivalent to a core cross-linked star polymer. The single-chain collapse process translates the molecular information written on the 1D graft copolymer into the 3D globular polymer nanoparticle, like protein folding. Unprecedented control over structural parameters was achieved, including the length, number, and composition of the side chains as well as cross-linking density. Different side chains formed distinct subdomains in the sterically congested nanoparticle state and further self-assembled into micellar aggregates in a selective solvent. Both experimental observations and computational simulations indicated that preorganization of the side chains in the block sequence produces subdomains which primarily follow the backbone length scale, while random sequences showed side chain-dependent scaling. Polymer nanoparticles with discrete multiple subdomains were produced by folding of the ternary block graft copolymers. Drastic differences in the self-assembly behavior of ABC- and ACB-sequenced nanoparticles indicate that the spatial organization of subdomains can be achieved by sequence control.
Synthesizing nanoporous polymer from the block polymer template by selective removal of the sacrificial domain offers straightforward pore size control as a function of the degree of polymerization (N). Downscaling pore size into the microporous regime (<2 nm) has been thermodynamically challenging, because the low N drives the system to disorder and the small-sized pore is prone to collapse. Herein, we report that maximizing cross-linking density of a block polymer precursor with an increased interaction parameter (χ) can help successfully stabilize the structure bearing pore sizes of 1.1 nm. We adopt polymerization-induced microphase separation (PIMS) combined with hyper-cross-linking as a strategy for the preparation of the bicontinuous block polymer precursors with a densely cross-linked framework by copolymerization of vinylbenzyl chloride with divinylbenzene and also Friedel–Crafts alkylation. Incorporating 4-vinylbiphenyl as a higher-χ comonomer to the sacrificial polylactide (PLA) block and optimizing the segregation strength versus cross-linking density allow for further downscaling. Control of pore size by N of PLA is demonstrated in the range of 9.9–1.1 nm. Accessible surface area to fluorescein-tagged dextrans is regulated by the relative size of the pore to the guest, and pore size is controlled. These findings will be useful for designing microporous polymers with tailored pore size for advanced catalytic and separation applications.
Surface modification offers an efficient and economical route to installing functional groups on a polymer surface. This work demonstrates that primary amine groups can be introduced onto a polymer surface via Buchwald–Hartwig amination, and the functionalized substrates can be chemically bonded to produce functional microfluidic devices. By activating the C-Cl bond in commercially used poly(chloro-p-xylylene) (parylene C) by Pd catalystand substituting Cl with the amine source, the amine groups are successfully
installed in a facile and recyclable manner. The substrates can be covalently bonded with each other via amine-isocyanate chemistry, providing much higher bonding strength compared to previous methods based on noncovalent adhesive coatings. As a result, transparent and flexible microfluidic channels can be fabricated that are compatible with organic solvents and high pressure. Retention of amine group reactivity in the channel suggests the potential of this methodology for the surface immobilization of functional
molecules for microfluidic reactors and biosensors.
A hierarchically porous polymer (HPP) consisting of micropores (∼1 nm) within a 3D continuous mesoporous wall (∼15 nm) was used to support well-defined Pt nanoparticles (2 nm in diameter) as a heterogeneous catalyst for the Suzuki–Miyaura cross-coupling reaction in the liquid phase. The ligand-capped nanoparticles were loaded into the polymer and treated with plasma to expose the active surface. The dual porosity was essential: the block polymer-templated mesopores provided the reactants facile access to the nanoparticle center, which was firmly immobilized by the microporous surface. Compared to inorganic mesoporous silica supports, which are intrinsically susceptible to basic hydrolysis, the Pt-HPP featured higher activity for all halide leaving groups, even in green solvents, as well as excellent recyclability. Only 5% decrease in activity was observed after 10 cycles. Pt-HPP was one of the most active heterogeneous catalysts for aryl chloride substrates compared to literature Pt or Pd examples.
We report the synthesis of thiopropionic acid-functionalized polymethacrylate-b-polystyrene (PMATA-b-PS) as a template for patterning Ag nanoarrays. Reversible addition-fragmentation chain transfer (RAFT) polymerization of silyl-protected propagyl methacrylate followed by chain extension with styrene produced a block copolymer precursor. Deprotection of the trimethylsilyl group and subsequent thiol-yne reaction afforded the target, PMATA-b-PS. While the entire series of the precursor was in the disordered phase, microphase separated morphologies were identified from PMATA-b-PS, indicating an increased interaction parameter (χ) as a result of introducing the acid groups. As a preliminary result, we show that an Ag nanoparticle array can be fabricated by selectively associating Ag+ ions to the PMATA cylinders in the thin film of PMATA-b-PS and removing the organic polymer layer by oxygen plasma treatment.
This study explores hyper-cross-linking of the cores of block copolymer micelles as a means to generate star polymers with a hyper-cross-linked core surrounded by linear corona arms. A solution of poly(methyl methacrylate)-b-polystyrene (MS) was prepared in acetonitrile to form micelles which were reacted with α,α’-dichloro-p-xylene in the presence of FeCl3 to produce core hyper-cross-linked star (CHS) polymers by selective cross-linking of the PS core. A kinetic investigation showed formation of high-molar mass species (>104 kg mol−1) within 1 h of reaction, which supported conversion of individual MS micelles into CHS polymers. We synthesized several CHS polymers by varying the PS core fractions from 20 to 53%. All the polymers possessed discrete spherical cores that were 19–60 nm in diameter and all were highly soluble in organic solvents retaining the CHS architecture. While permanent microporosity was not detected by gas sorption measurements, increased dye uptake of CHS polymer in solution suggests utility of CHS polymers as stable and solution-processible nanocontainers with accessible free volume in the core.
Fine-tuning and pore environment control of covalently connected metal–organic framework (MOF) and mixed-matrix membrane (MMM) composite materials were achieved. Core–shell-type, dual-functionalized, zirconium-based MOFs were prepared through a postsynthetic ligand exchange (PSE) process, and active vinyl functionalities on the surface of MOF nanoparticles were utilized for polymerization by forming interfacial-covalent connections between MOF nanoparticles and polymeric membranes via thiol–ene click photopolymerization. The target functionality of the MOF pore originated from the parent MOFs, allowing pore engineering of the MOF–MMM composite materials. A series of defect-free, interface-controlled, and core-functionalized MOF–MMMs were prepared through the present methodology, and the NO2-functionalized/covalently connected MOF–MMM showed the highest CO2 permeability and solubility without loss of selectivity. This facile and versatile approach will be useful for the fabrication of functional MOF nanoparticle-based membranes for various applications, such as catalysis and separation.
Metal catalysts are generally supported on hard inorganic materials because of their high thermochemical stabilities. Here, we support Pd catalysts on a thermochemically stable but “soft” engineering plastic, polyphenylene sulfide (PPS), for acetylene partial hydrogenation. Near the glass transition temperature (~353 K), the mobile PPS chains cover the entire surface of Pd particles via strong metal-polymer interactions. The Pd-PPS interface enables H2 activation only in the presence of acetylene that has a strong binding affinity to Pd and thus can disturb the Pd-PPS interface. Once acetylene is hydrogenated to weakly binding ethylene, re-adsorption of PPS on the Pd surface repels ethylene before it is further hydrogenated to ethane. The Pd-PPS interaction enables selective partial hydrogenation of acetylene to ethylene even in an ethylene-rich stream and suppresses catalyst deactivation due to coke formation. The results manifest the unique possibility of harnessing dynamic metal-polymer interaction for designing chemoselective and long-lived catalysts.
Perovskite nanocrystals are promising luminescent materials with synthetic feasibility and band gap tunability. Nonetheless, application of the perovskite nanocrystals to light-emitting devices has been challenging because of the intrinsic poor colloidal stability and environmental vulnerability issues. Here, we introduce a new protocol for highly air-stable perovskite nanocrystal layers with a tunable band gap via a simple nanocrystal pinning process. The nanocrystals were composed of CH3NH3PbBr3 (MAPbBr3) mixed with (vinylbenzylamine)2PbBr4 ((VBzA)2PbBr4), which contains a photopolymerizable structure-directing ligand. Along with the compostion of (VBzA)2PbBr4, the band gap of the perovskite layer continuously increased with the reduction of the nanocrystal size and also lattice distortion. The nanocrystal film readily polymerized upon exposure to visible light was highly stable under humid air more than 15 days. Its application to bluish-green light-emitting diodes is demonstrated.
We report polymerization-induced self-assembly via controlled cross-linking copolymerization to produce robust block copolymer micelles with spherical, elongated, and branched shapes. Reversible addition–fragmentation chain transfer (RAFT) copolymerization of styrene and divinylbenzene (DVB) or 1,2-bismaleimidoethane (BMI) as a cross-linker in the presence of a polylactide macro-chain transfer agent (PLA-CTA) was performed in acetonitrile, which is a non-solvent to polystyrene (PS). The addition of the cross-linker accelerates the copolymerization compared to styrene homopolymerization, which leads to the formation of block polymer micelles within a shorter time frame, followed by in situ inter-chain cross-linking. The micelles are virtually identical to the core cross-linked star polymer consisting of a cross-linked polystyrenic core surrounded by a PLA corona. Molecular weights up to more than 1000 kg mol−1 could be obtained with relatively narrow dispersity values (1.1–1.4). In the case of copolymerization with DVB, the micellar morphology changes from spherical to elongated and branched shapes with increasing conversion. The size and morphology of the micelles are retained in a good solvent to PS, suggesting that the in situ cross-linking effectively stabilizes the micellar core. BMI undergoes alternating copolymerization with styrene in the early stage of polymerization and yields spherical micelles exclusively, because the densely cross-linked core seems to prevent further morphological transition.
Mesoporous nonoxide ceramics are attractive for applications such as catalytic supporters and separations with exceptional thermochemical stability. Here we report on the one-step preparation of microphase-separated bicontinuous organic–inorganic polymer precursors for forming 3D continuous polymer-derived ceramic monoliths without an external block copolymer template and annealing steps. We combined polymerization-induced phase separation with in situ hybrid block polymer formation from a mixture of a preceramic monomer, a cross-linker, and a thermally decomposable organic segment containing a terminal chain transfer agent. The resultant cross-linked polymeric monoliths, moldable to any desired shape, were converted to 3D-continuous mesoporous silicon carbonitride ceramics with a pore size in the 3–11 nm range and a surface area of 107–410 m2 g–1 by varying the molar mass of the sacrificial organic block and the pyrolytic temperature. The 3D-disordered pore structure is beneficial for retaining the monolithic shape via isotropic shrinkage during ceramization. The distinctive characteristics of this synthetic approach, which are the absence of a solvent, a structure-directing block copolymer, and an annealing process, are affordable for the large production of nanoporous ceramic monoliths for various high-temperature applications and should be applicable for additive manufacturing with direct polymerizability for the fabrication of hierarchically porous materials in complex shapes with dimensional scalability.
We developed a block polymer-based synthetic route to sulfonated porous composites (SPCs) with precisely controlled nanopore size. By reducing the pore size to <4 nm and introducing a high density of surface sulfonic acid, the permeation of vanadium ions was effectively suppressed. We employed a polymerization-induced microphase separation (PIMS) process, in which a polyethylene fiber mat impregnated with a liquid polymerization mixture was spontaneously transformed into a fiber-reinforced and cross-linked block polymer membrane. Selective etching and sulfonation then produced the target composite membrane. In a vanadium redox flow battery (VRFB) cell, an SPC with 3.6 nm-sized mesopores, 109 m2 g–1 of specific surface area, and 0.3 mL g–1 of mesoporosity outperformed a Nafion 212 membrane of similar thickness, providing higher proton conductivity and much lower vanadium permeability. Thanks to the composite reinforcement, the membrane demonstrated remarkably enhanced mechanical stability. The SPC membrane could be successfully operated up to 300 cycles. Compared with Nafion 212, the SPC exhibited higher energy efficiencies (EEs) and higher discharge capacity retention. These results suggest the promise of block polymer-based permselective membranes in advanced battery applications.
We report that an alternating semifluorinated copolymer of chlorotrifluoroethylene (CTFE) and vinyl ether (VE) is an attractive platform for the synthesis of heterograft copolymers consisting of two distinct side chains. The radical terpolymerization of CTFE with PLA-tethered vinyl ether (PLAVE) synthesized by ring-opening polymerization and isobutyl vinyl ether (IBVE) as a spacer produced PLA-grafted fluorinated copolymer via a “grafting-through” manner. Two PLAVEs with different molar masses (2 and 10 kg mol–1) were successfully incorporated, and the grafting density could be controlled by varying the [PLAVE]/[IBVE] initial molar ratio. From the chlorine atoms in the CTFE repeating units, atom transfer radical polymerization (ATRP) of styrene was further employed to grow PS side chains following a “grafting-from” mechanism per each (CTFE-alt-VE) repeating unit dyad. First-order kinetics was observed for the styrene polymerization and supported controlled growth of PS. The resulting heterograft copolymers possessed regularly spaced PS chains and statistically distributed PLA chains on the backbone, generating a nanoscopic disordered morphology via microphase separation driven by incompatibility between PLA and PS. By copolymerization of styrene and divinylbenzene (DVB) in neat ATRP condition, a cross-linked polymer monolith with the disordered bicontinuous morphology could be also prepared via polymerization-induced microphase separation. The cross-linked precursor was converted into a mesoporous polymer with pore size of 3.7–10.4 nm by removal of PLA. The mesopore size was tunable by adjusting the PLA molar mass and styrene/DVB molar ratio.
Few studies aiming to develop a glue with an underwater reusable adhesive property have been reported because combining the two properties of reusable adhesion and underwater adhesion into a single glue formulation is a challenging issue. Herein, preparation of a simple mixture of poly(vinyl alcohol) (PVA) and a well-known phenolic compound, namely, tannic acid (TA), results in an underwater glue exhibiting reusable adhesion. We named the adhesive VATA (PVA + TA). Using VATA, two stainless steel objects (0.77 kg each) are able to be instantly attached. In addition to the high adhesive strength, surface-applied VATA in water retains its adhesive capability even after 24 h. In contrast, cyanoacrylate applied under the same water condition rapidly loses its adhesive power. Another advantage is that VATA’s adhesion is reusable. Bonded objects can be forcibly detached, and then the detached ones can be reattached by the residual VATA. VATA maintains nearly 100% of its initial adhesive force, even after 10 repetitions of attach–detach cycles. VATA bonds various materials ranging from metals and polymers to ceramics. Particularly, we first attempt to test the toxicity of the underwater adhesives using an invertebrate nematode, Caenorhabditis elegans and gold fish (vertebrate) due to potential release to the environment.
Redox-active polytriarylamine with hydroxyl groups is a useful material for optoelectronic applications, especially in the solution-processable multilayer devices. A novel regiocontrolled triarylamine-based polymer, poly(di-5-naphthyl-2-ol)phenylamine, with 2-naphthol units was synthesized via oxidative coupling polymerization. Polymerization in tetrahydrofuran using a Cu-amine complex oxidant under O2 atmosphere produced polymers with number-averaged molecular weights as high as 11,300 g mol−1. The structure of the polymer was characterized by 1H and 13C NMR spectroscopy, showing that the oxidative coupling polymerization occurred at the outer ortho position of the 2-naphthols, preserving the hydroxyl groups. The polymer exhibited good solubility in polar aprotic solvents, with a high thermal stability of 446 °C that corresponded to 5% weight loss. The UV–vis absorption of the polymer was similar to that of DNPA, indicating that the kinked-structured polymer hindered the formation of charge-transfer complexes. These results suggest promising applications of the developed polymer in optoelectronic devices.
Poly(biphenylene oxide)s (PBPOs) containing two pendent trifluoromethyl groups were synthesized from AB-type monomers, 4ʹ-hydroxy-4-fluoro-3,5-bis(trifluoromethyl)biphenyl and its 3ʹ-hydroxyl isomer. The displacement reaction of fluorine leaving group activated by the two trifluoromethyl groups at the ortho-positions produced high-molecular-weight polymers with Mn up to 101,000 g/mol, indicating the nucleophilic aromatic substitution reaction proceeded effectively. PBPOs with para-, meta-, and mixed ether linkages were obtained and well characterized by FTIR and 1H/19F NMR spectroscopies. All PBPOs were amorphous and soluble in a wide range of organic solvents, and exhibited even more enhanced thermal stability than the previously reported two meta-trifluoromethyl substituted analogues. Increasing the para-linkage fraction in the polymer generally improved solubility and increased Tg in contrast to the meta-trifluoromethyl case, where para-linked polymer was poorly soluble and semicrystalline. This suggests that the ortho-trifluoromethyl substituents are more effective for the synthesis of para-linked PBPOs. They also showed low refractive indices and birefringence values.
We report the synthesis of a series of statistical terpolymer poly[(methyl methacrylate)-co-lauryl methacrylate-co-2-((3,5-bis(4-carbamoyl-3-(trifluoromethyl)phenoxy)benzyloxy)carbonylamino)ethyl methacrylate] (P(MMA-co-LMA-co-BMA)) by reversible addition–fragmentation chain transfer polymerization and their aggregation behaviors in solution. In toluene, the solution behavior of terpolymer was controlled by the molar fractions of lauryl methacrylate (LMA) and benzamide-containing methacrylate (BMA) in the polymer, which increased solubility and promoted hydrogen bonding between the primary aromatic amides, respectively. Temperature-dependent 1H NMR spectroscopy also indicated gradual dissociation of the hydrogen bonds with increasing temperature. For the polymer containing 2.7 mol % of LMA and 2.7 mol % of BMA repeating units, we demonstrated that dissolving the polymer in tetrahydrofuran as a good solvent and switching the solvent with toluene produced polymer nanoparticles with diameters of several tens of nanometers, as observed by dynamic light scattering. Intramolecular hydrogen bonding was dominant and induced the noncovalent chain collapse. When the temperature of the particle dispersion in toluene at a concentration > 30 mg/mL was increased from RT to 50 °C, a significant increase in viscosity was observed. This behavior was not observed in a toluene solution of poly(methyl methacrylate), which showed decreased viscosity at a higher temperature. The viscosity increase was accompanied by a decrease in the particle size, and both were attributed to the dissociation of some intramolecular hydrogen bonds within the particles, which can increase the number of individual chains in toluene and result in more intermolecular interactions.
We report the self-assembly of monolayer vesicles from Janus core–shell bottlebrush polymers. A route was developed to synthesize doubly grafted bottlebrush copolymers (DGBCPs) possessing A-b-B and B′-b-C side chains on a single repeating unit. Graft-through ring-opening metathesis polymerization of a norbornene moiety installed by single unit monomer insertion allowed us to place the backbone on any repeating unit of the core (B and B′) block. By decorating each core chain end with different chains via reversible addition–fragmentation chain transfer polymerization, we can obtain nanoobjects with an asymmetric B core and a phase-separated A/C shell. We demonstrate that polystyrene-branch-polystyrene′ and polylactide-b-polystyrene-branch-polystyrene′-b-poly(n-butyl acrylate) macromonomers can be successfully synthesized and polymerized to produce DGBCPs in high yields (81–94% conversion) with an absolute molar mass of 149–395 kg mol–1 and a dispersity of 1.18–1.38. In a solvent slightly more selective to A than C, self-assembly of monolayer vesicles with diameter of <100 nm was observed by transmission electron microscopy. Dissipative particle dynamics simulations suggest that increasing the backbone length and moving the backbone toward the B′/C interface increases the backbone bending energy and favors a lower curvature. The spontaneous curvature appears to prefer a particular layer radius, avoiding bilayer formation.
We report a route to synthesize polylactide-b-poly(ether sulfone)-b-polylactide (PLA-b-PES-b-PLA) containing PES and PLA, which provide a mechanically robust framework and a sacrificial template for pore formation, respectively. High-molar mass PES terminated with fluorine groups was synthesized by the step-growth nucleophilic aromatic substitution (SNAr) reaction, and the chain ends were transformed into benzylic hydroxyl groups by chain end modification. Growth of the PLA using the hydroxyl groups as initiating sites via chain-growth ring opening transesterification polymerization (ROTEP) produced the target triblock copolymer. Although the step-growth polymerization produced a PES middle block with high dispersity, small angle X-ray scattering (SAXS) and transmission electron microscopy (TEM) analyses indicated the formation of an ordered lamellar morphology. We further demonstrated that a nanoporous PES with slit-like pores could be obtained by selective removal of the PLA.
Heteroarm core cross-linked star (CCS) polymers consist of two different polymer chains covalently joined to a cross-linked core. We investigated their self-assembly behavior to understand whether intramolecular segregation can be induced during synthesis, to produce spatial domains enriched with each polymer, and whether they would exhibit well-defined microphase separation morphologies as a result. Heteroarm CCS polymers containing polylactide (PLA) and polystyrene (PS) arms were synthesized by reversible addition–fragmentation chain transfer copolymerization of styrene and 1,2-bis(maleimidoethane) in the presence of a PLA-macro chain transfer agent (PLA-CTA), followed by chain extension with styrene (the in–out route). Dynamic light scattering, transmission electron microscopy, and small angle X-ray scattering analyses were employed to examine the self-assembly behavior in toluene and acetonitrile, as a relatively neutral and a PLA-selective solvent, respectively. Above a critical PS molar mass, lamellar-like and spherical morphologies were observed, formed by microphase separation into discrete PLA and PS domains. The increase in order with increasing PS molar mass was consistent with the segregation strength-dependent microphase separation behavior. In contrast, when the CCS polymer was synthesized by simultaneously joining PLA and PS chains (the multi macroinitiatior route) it produced rather ill-defined self-assemblies, suggesting that styrene chain extension via the in–out process is important to achieve intramolecular segregation. Using the more PLA-selective acetonitrile as a polymerization solvent indeed produced more well-defined supermicelles with PS cores and PLA coronas, confirming that intramolecular segregation can be driven by the incompatibility of the growing PS to the intramolecular environment, including PLA and the solvent.
We investigated the influence of heavy halogen atoms (Br and I) on xanthene dyes for polymerization based on visible-light photoredox initiation. Since the heavy atoms directly affect intersystem crossing (ISC), which can act as a gatekeeper in the photoredox cycle and which was expected to also affect intermolecular photoinduced electron transfer (PET), we attempted to quantify the influence of the halogens. Six different xanthene dyes were chosen based on the number and types of heavy atoms on the xanthene ring. Thus, the photopolymerization degree clearly increased in the following order: fluorescein < 4′,5′-dibromofluorescein ≤ 2′,4′,5′,7′-tetrabromofluorescein < 2′,4′,5′,7′-tetraiodofluorescein. Furthermore, 4′,5′-dibromorhodamine 6G showed a drastic enhancement in the photopolymerization degree, compared with rhodamine 6G. Therefore, we concluded that the presence of halogens on the xanthene ring increases the photoredox initiating performance due to the enhanced ISC efficiency and PET rate.
We investigated the microphase separation behavior of well-defined poly(arylene ether sulfone)-b-polylactide (PES-b-PLA) diblock copolymers. PES was synthesized by the nucleophilic aromatic substitution polymerization of 4-fluoro-4′-hydroxydiphenyl sulfone potassium salt in the presence of an allyl-functionalized initiator, which follows a chain growth condensation polymerization mechanism. A hydroxyl group installed via a thiol-ene reaction was utilized as the initiating site for the ring opening polymerization of d,l-lactide, producing the target polymer. The polymers were further purified by preparative size-exclusion chromatography and analyzed by small-angle X-ray scattering with temperature variations from room temperature to 150 °C. The PES block was glassy in the employed temperature range, but the PLA chains provided sufficient mobility for ordering of the block copolymer when PES was the minor fraction. An order-disorder transition (ODT) with changing temperature could not be located because PLA was not stable above 170 °C. From the degree of polymerization values of the polymers near the ODT, the Flory–Huggins interaction parameter, χ, could be roughly estimated as 0.12 at 150 °C. This high χ value suggests that engineering plastic-containing block copolymers could be useful in advanced lithographic and filtration applications.
We propose the defunctionalization of vinyl polymers as a strategy to access previously inaccessible polyolefin materials. By utilizing B(C6F5)3-catalyzed deoxygenation in the presence of silane, we demonstrate that eliminating the pendent ester in poly(methyl acrylate) effectively leaves a linear hydrocarbon polymer with methyl pendants, which is polypropylene. We further show that a polypropylene-b-polystyrene diblock copolymer and a polystyrene-b-polypropylene-b-polystyrene triblock copolymer can be successfully derived from the poly(methyl acrylate)-containing block polymer precursors and exhibit quite distinct materials properties due to their chemical transformation. This unique postpolymerization modification methodology, which goes beyond the typical functional group conversion, can offer access to a diverse range of unprecedented polyolefin block polymers with a variable degree of functional groups.
We report self-assembly of a statistical copolymer poly(styrene-co-methyl methacrylate) (P(S-co-MMA)) containing ionic liquid (IL)-philic methyl methacrylate (MMA) and IL-phobic styrene (S) repeating units in IL for fabrication of electrolyte-gated organic transistors. P(S-co-MMA)s with high MMA contents were synthesized by copolymerization of styrene and MMA via a reversible addition–fragmentation chain transfer (RAFT) process, and their behavior in 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([EMI][TFSI]) was investigated. While dynamic light scattering analysis showed formation of a micellar solution at low concentration, the elastic modulus of the viscoelastic solution increased significantly more than the loss modulus at high concentration. Small angle X-ray scattering analysis suggested ill-defined phase separation between PS-rich segments and PS-lean segments swollen in [EMI][TFSI]. The resulting P(S-co-MMA)/[EMI][TFSI] mixture exhibited increased ionic conductivity compared to the PS-b-PMMA-b-PS block polymer gel, as well as superior device performance in transistor gating experiments.
We report the synthesis of microporous hyper-cross-linked polymers (HCPs) with increased specific surface area and porosity by the in situ removal of trimethylsilyl (TMS) groups during hyper-cross-linking. We synthesized poly(4-trimethylsilylstyrene-co-vinylbenzyl chloride-co-divinylbenzene)s (P(TMSS-co-VBzCl-co-DVB)s) with different compositions by reversible addition–fragmentation chain transfer copolymerization and converted them into HCPs by reacting with FeCl3 in 1,2-dichloroethane. The nearly quantitative removal of the TMS groups was observed during the reaction following the electrophilic aromatic substitution mechanism, where the TMS group shows higher reactivity than an aromatic hydrogen. Substantial enhancement in pore characteristics including surface area, microporosity, and mesoporosity was noticed up to a certain level of TMSS incorporation, compared with HCP derived from P(VBzCl-co-DVB). We suggest the porogenic TMS group increases porosity mainly by in situ removal via facilitated substitution reaction, which creates permanent voids in the hyper-cross-linked network. The use of TMSS provides a feasible and complementary route to tuning the pore characteristics of HCPs by varying DVB content, and is applicable to the synthesis of hierarchically porous polymers containing micropores within a mesoporous framework from block polymer precursors.
We report a new methodology that allows for forming micropores in hierarchically porous polymers by employing the reversible addition–fragmentation chain transfer (RAFT) copolymerization of conjugated multi-vinyl cross-linkers with styrene. Using divinylbenzene, 4,4′-divinylbiphenyl, 1,3,5-tris(4-vinylphenyl)benzene and tetrakis(4-vinylbiphenyl)methane as cross-linkers, the RAFT copolymerization was carried out in the presence of polylactide macro-chain transfer agents. During the polymerization, microphase separation occurred spontaneously to produce cross-linked block polymer precursors with a bicontinuous morphology composed of polylactide and cross-linked polystyrene microdomains. Hierarchically porous polymers with strong fluorescence were successfully derived by polylactide etching. We demonstrate that the rigid conjugated structure of the cross-linkers with a high cross-linking density is critical for creating the micropores and for stabilizing the mesopores that are templated by the polylactide domain.
We investigated proton conductivity and the permeability of monovalent cations across sulfonated mesoporous membranes (SMMs) prepared with well-defined pore sizes and adjustable sulfonic acid content. Mesoporous membranes with three-dimensionally continuous pore structure were produced by the polymerization-induced microphase separation (PIMS) process involving the reversible addition–fragmentation chain transfer (RAFT) copolymerization of styrene and divinylbenzene in the presence of a polylactide (PLA) macrochain transfer agent and subsequent PLA etching. This allowed us to control pore size by varying PLA molar mass. Postsulfonation of the mesoporous membranes yielded SMMs whose pore structure was retained. The sulfonic acid content was adjusted by reaction time. While proton conductivity increased with increasing ion exchange capacity (IEC) without noticeable dependence on the pore size, ion permeability was strongly influenced by the pore size and IEC values. Decreasing pore size and increasing IEC resulted in a decrease in ion permeability, suggesting that ions traverse across the membrane via the vehicular mechanism, through the mesoporous spaces filled with water. We further observed that the permeability of the vanadium oxide ion was dramatically suppressed by reducing the pore size below 4 nm, which was consistent with preliminary vanadium redox flow battery data. Our approach suggests a route to developing permselective membranes by decoupling proton conductivity and ion permeability, which could be useful for designing separator materials for next-generation battery systems.
Kim, Sun Dal; Lee, Byungyong; Byun, Taejoon; Chung, Im Sik; Park, Jongmin; Shin, Isaac; Ahn, Nam Young; Seo, Myungeun; Lee, Yunho; Kim, Yeonjoon; Kim, Woo Youn; Kwon, Hyukyun; Moon, Hanul; Yoo, Seunghyup; Kim, Sang Youl
The key component currently missing for the next generation of transparent and flexible displays is a high-performance polymer material that is flexible, while showing optical and thermal properties of glass. It must be transparent to visible light and show a low coefficient of thermal expansion (CTE). While specialty plastics such as aromatic polyimides are promising, reducing their CTE and improving transparency simultaneously proved challenging, with increasing coloration the main problem to be resolved. We report a new poly(amide-imide) material that is flexible and displays glass-like behavior with a CTE value of 4 parts per million/°C. This novel polymer was successfully used as a substrate to fabricate transparent and flexible indium-gallium-zinc oxide thin-film transistors.
A series of well-defined poly(arylene ether sulfone)s (PESs) as a rod-type block was synthesized by chain-growth condensation polymerization from a diphenyl sulfone-type initiator containing a fluorine leaving group and an allyl moiety. Interestingly, these oligomeric PESs exhibited lower critical solution temperature (LCST)-type phase transition behavior in organic solvents, i.e., 1,2-dimethoxyethane (DME) and chloroform. The clouding point temperature was affected by the molecular weight and concentration of the polymers. The cloud temperature decreased as the molecular weight polymers and the concentration of polymer solution increased. And also two series of rod-coil type poly(arylene ether sulfone)-b-polylactides were synthesized by controlled ring-opening esterification polymerization of dl-lactide with a PES-derived macroinitiator in which the allyl group was transformed into an aliphatic hydroxyl group by a thiol-ene click reaction. These diblock copolymers also exhibited LCST behavior in DME, and the nanoscale size of the aggregates increased upon heating.
Soluble and transparent poly(amide‐imide)s (PAIs) with a glass transition temperature over 300 °C were synthesized from the alicyclic diacid monomers containing a biphenyl unit with two trifluoromethyl (CF3) groups. Combination of two isomeric biphenyl units with CF3 groups significantly improves the glass transition temperature of the corresponding PAIs. The increase of unsymmetrical biphenyl units increased the glass transition temperature of the polymers by reducing the average chain packing distance.
The branching point of the side‐chain of naphthalenediimide (NDI)‐based conjugated polymers is systematically controlled by incorporating four different side‐chains, i.e., 2‐hexyloctyl (P(NDI1‐T)), 3‐hexylnonyl (P(NDI2‐T)), 4‐hexyldecyl (P(NDI3‐T)), and 5‐hexylundecyl (P(NDI4‐T)). When the branching point is located farther away from the conjugated backbones, steric hindrance around the backbone is relaxed and the intermolecular interactions between the polymer chains become stronger, which promotes the formation of crystalline structures in thin film state. In particular, thermally annealed films of P(NDI3‐T) and P(NDI4‐T), which have branching points far away from the backbone, possess more‐developed bimodal structure along both the face‐on and edge‐on orientations. Consequently, the field‐effect electron mobilities of P(NDIm‐T) polymers are monotonically increased from 0.03 cm2 V−1 s−1 in P(NDI1‐T) to 0.22 cm2 V−1 s−1 in P(NDI4‐T), accompanied by reduced activation energy and contact resistance of the thin films. In addition, when the series of P(NDIm‐T) polymers is applied in all‐polymer solar cells (all‐PSCs) as electron acceptor, remarkably high‐power conversion efficiency of 7.1% is achieved along with enhanced current density in P(NDI3‐T)‐based all‐PSCs, which is mainly attributed to red‐shifted light absorption and enhanced electron‐transporting ability.
We developed a facile methodology for fabricating a free-standing mixed-matrix membrane (MMM) containing covalently incorporated metal–organic framework (MOF) particles up to 60 wt% by utilizing thiol–ene photopolymerization with the MOF consisting of vinyl functionality. Vinyl-functionalized UiO-66 (UiO-66-CH[double bond, length as m-dash]CH2) was synthesized from 2-vinyl-1,4-dicarboxylic acid with ZrCl4, and a free-standing MMM was readily produced by irradiation of a polymerization mixture containing UiO-66-CH[double bond, length as m-dash]CH2, poly(ethylene glycol) divinyl ether (PEO-250), pentaerythritol tetra(3-mercaptopropionate) (PETM), 2,2′-(ethylenedioxy)diethanethiol (EDDT), and 2,2-dimethoxy-2-phenylacetophenone (DMPA) as a photoradical initiator. Assorted analyses combining FTIR, thermogravimetric analysis, scanning electron microscopy, energy dispersive X-ray spectroscopy, and X-ray diffraction strongly supported the fact that the desired MMM containing well-dispersed UiO-66-CH[double bond, length as m-dash]CH2 particles was successfully produced by C–S bond formation, which provided strong union of the MOF with the polymer matrix without interfacial voids. The produced MMM was highly flexible and showed improved mechanical properties as compared to the pristine polymeric membrane, indicating that the covalently immobilized UiO-66-CH[double bond, length as m-dash]CH2 particles were homogeneously distributed in the polymer matrix. Gas permeability across the MMM was significantly enhanced compared with the pristine polymeric membrane as diffusion of the gas molecules was facilitated in the porous space in the MOF.
We report a blending mechanism of polystyrene-b-poly(ethylene oxide) (PS-b-PEO) and PS homopolymer (homoPS) at the air/water interface. Our blending mechanism is completely different from the well-known “wet–dry brush theory” for bulk blends; regardless of the size of homoPS, the domain size increased and the morphology changed without macrophase separation, whereas the homoPS of small molecular weight (MW) leads to a transition after blending into the block copolymer domains, and the large MW homoPS is phase-separated in bulk. The difference in blending mechanism at the interface is attributed to adsorption kinetics at a water/spreading solvent interface. Upon spreading, PS-b-PEO is rapidly adsorbed to the water/spreading solvent interface and forms domain first, and then homoPS accumulates on them as the solvent completely evaporates. On the basis of our proposed mechanism, we demonstrate that rapid PS-b-PEO adsorption is crucial to determine the final morphology of the blends. We additionally found that spreading preformed self-assemblies of the blends slowed down the adsorption, causing them to behave similar to bulk blends, following the “wet–dry brush theory”. This new mechanism provides useful information for various block copolymer-homopolymer blending systems with large fluid/fluid interfaces such as emulsions and foams.
We report the preparation of hierarchically porous polymers containing fully interconnected and controlled micro-, meso-, and macropores, where a hyper-cross-linked microporous polymer skeleton forms a reticulating mesoporous wall that supports a highly porous macropore framework. These materials provide high specific surface area and >90% porosity, useful for rapid sorption of organic molecules.
Recently, because of rapid advances in electrical vehicles, unmanned air vehicles, and humanoid mobile robots, structural energy storage devices with a concurrent capability to store electrochemical energy and to support mechanical loads have been in the spotlight. However, a big hurdle to realizing an integrated electro-chemo-mechanical system is to develop highly compatible active electrodes and structural electrolytes with superior mechanical strength and electrochemical functionality while retaining light weight. We report a load-bearing structural supercapacitor by utilizing a bicontinuous PEO-b-P(S-co-DVB) structural electrolyte and carbon-coated Ni-Co nanowires grown on carbon fiber woven fabric. A liquid polymerization mixture between the electrodes is transformed into a solid-state block copolymer electrolyte, preserving conformal contact with the nanostructured electrode surface. The polymerization-induced microphase separation produces a bicontinuous morphology of cross-linked hard domain and liquid-like conductive domain in the electrode, providing high modulus and high conductivity. The resulting structural supercapacitor is able to operate under tensile and even bending load, suggesting its wide potential applications.
Herein, a study on a new lower critical solution temperature (LCST) polymer in an organic solvent by an electrochemical technique has been reported. The phase-transition behavior of poly(arylene ether sulfone) (PAES) was examined on 1,2-dimethoxyethane (DME). At a temperature above the LCST point, polymer molecules aggregated to create polymer droplets. These droplets subsequently collided with an ultramicroelectrode (UME), resulting in a new form of staircase current decrease. The experimental collision frequency and collision signal were analyzed in relation to the concentration of the polymer. In addition, the degree of polymer aggregation associated with temperature change was also observed.
This image from the research of Soobin Kim and Myungeun Seo on page 900 shows a scanning electron micrograph of a hierarchically porous polymer synthesized by combination of hyper‐crosslinking with polymerization‐induced microphase separation (PIMS). Three‐dimensionally continuous mesopores with size of ca. 10 nm are evident. The PIMS process allows them to readily produce a crosslinked block polymer precursor with a disordered bicontinuous morphology composed of polylactide (PLA) and polystyrenic microdomains. A hyper‐crosslinking reaction degrades the PLA to generate the mesoporous space, and it simultaneously creates micropores smaller than 2 nm (not visible) within the polystyrenic microdomain to yield the hierarchical pore structure. This provides improved stability and accelerated diffusion to microporous surface. (DOI: 10.1002/pola.28966)
Microcapsules with nanoporous membranes can regulate transmembrane transport in a size-dependent fashion while protecting active materials in the core from the surrounding, and are thereby useful as artificial cell models, carriers for cells and catalysts, and microsensors. In this work, we report a pragmatic microfluidic approach to producing such semipermeable microcapsules with precise control of the cutoff threshold of permeation. Using a homogeneous polymerization mixture for the polymerization-induced microphase separation (PIMS) process as the oil phase of water-in-oil-in-water (W/O/W) double emulsions, a densely cross-linked shell composed of a bicontinuous nanostructure that percolates through the entire thickness is prepared, which serves as a template for a monolithic nanoporous membrane of microcapsules with size-selective permeability. We demonstrate that the nanopores with precisely controlled size by the block polymer self-assembly govern molecular diffusion through the membrane and render manipulation of the cutoff threshold.
We report on the phase separation behaviors of polymerization mixtures containing a polylactide macro-chain transfer agent (PLA-CTA), styrene, divinylbenzene, hydroxyl-terminated PLA (PLA-OH), and a molecular chain transfer agent which enable the ability to tune the pore size of a cross-linked polymer monolith in a facile manner. Cross-linked monoliths were produced from the mixtures via reversible addition-fragmentation chain transfer (RAFT) polymerization and converted into cross-linked porous polymers by selective removal of PLA while retaining the parent morphology. We demonstrate that pore sizes are tunable over a wide range of length scales from the meso- to macroporous regimes by adjusting the ratio of PLA-CTA to PLA-OH in the reaction mixture which causes the phase separation mechanism to change from polymerization-induced microphase separation to polymerization-induced phase separation. The possibility of increasing porosity and inducing simultaneous micro- and macrophase separation was also realized by adjustments in the molar mass of PLA which enabled the synthesis of hierarchically meso- and macroporous polymers.
본 발명은 블록 공중합체 자기조립을 응용하여 세공 크기가 정밀하게 조절된 다공성 고분자막을 제조할 수 있는 한외여과막용 블록공중합체 및 이의 제조방법에 관한 것이다.
본 발명의 블록공중합체는 블록공중합체를 형성하는 고분자들의 분자량과 함량을 조절하여 세공 크기와 분포를 정밀하게 제어할 수 있으며, 또한, 현재 한외여과막으로 사용되고 있는 폴리이서술폰을 기본 소재로 사용하고 있어 기계적 물성이 우수하다.
본 발명은 현재 한외여과 분리막에 쓰이고 있는 폴리이서술폰을 포함하는 블록 공중합체를 합성하고 이에 기반한 나노다공성 고분자 제조 기술 개발을 통해 차세대 한외여과용 나노다공성 여과막을 구현하였으며, 기존의 상반전법을 통한 폴리이서술폰 한외여과 분리막 제조공정을 적용할 수 있어 평판 분리막 또는 중공사막 분리막으로 쉽게 가공할 수 있으므로 높은 산업적 응용 가치를 가진다.
We explored reversible addition-fragmentation chain transfer (RAFT) copolymerization of 1,2-bis(maleimidoethane) (BMI) with styrene (S) in the presence of polylactide macro-chain transfer agent (PLA-CTA) as a means to synthesize heteroarm core cross-linked star (CCS) polymers consisting of PLA and PS arms (PLAnPSn). Because of the strong alternating tendency of maleimide and styrenic double bonds, copolymerization of BMI with an excess of S depleted BMI in the early stage of polymerization forming a cross-linked core. The remaining S was successively polymerized to grow PS arms from the core, completing PLAnPSn via “in–out” mechanism. Use of a stoichiometric amount of S produced PLAn, which could be used as a macro-CTA for the synthesis of more well-defined PLAnPSn. Compared with divinylbenzene, copolymerization of BMI with S was much more effective for core formation suggesting the importance of the alternating character of the copolymerization. While PLAnPSn existed as stable nanoparticles in a neutral solvent in contrast to linear PLA-b-PS, it also self-assembled to form microphase-separated structures in a selective solvent and in bulk indicating that PLA and PS arms can be intramolecularly segregated.
We report on the use of photoinitiated reversible addition–fragmentation chain transfer (RAFT) polymerization for the facile fabrication of cross-linked nanoporous polymer films with three-dimensionally (3D) continuous pore structure. The photoinitiated polymerization of isobornyl acrylate (IBA) in the presence of 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (CTA) and 2,2-dimethoxy-2-phenylacetophenone as a photoinitiator proceeded in a controlled manner, yet more rapidly compared to thermally initiated polymerization. When polylactide-macroCTA (PLA-CTA) was used, PLA-b-PIBA with high molar mass was obtained after several minutes of irradiation at room temperature. We confirmed that microphase separation occurs in the PLA-b-PIBA and that nanoporous PIBA can be derived from the PLA-b-PIBA precursor by selective PLA etching. To fabricate the cross-linked nanoporous polymer, IBA was copolymerized with ethylene glycol diacrylate (EGDA) in the presence of PLA-CTA to produce a cross-linked block polymer precursor consisting of bicontinuous PLA and P(IBA-co-EGDA) microdomains, via polymerization-induced microphase separation. We demonstrated that nanoporous P(IBA-co-EGDA) monoliths and films with 3D continuous pores can be readily obtained via this approach.
Partially sulfonated amphiphilic poly(arylene ether sulfone)s (PSPAESs) were synthesized by one-step nucleophilic aromatic substitution copolymerization. A 4-fluoro-4′-hydroxydiphenyl sulfone potassium salt was used as a hydrophobic monomer, and 5-((4-fluorophenyl)sulfonyl)-2-hydroxybenzenesulfonic acid as a hydrophilic monomer bearing a sulfonic acid group was synthesized from the hydrophobic monomer via selective sulfonation. 1H and 13C nuclear magnetic resonance spectroscopy analysis of PSPAESs indicated formation of statistical amphiphilic copolymers with control over the degree of sulfonation by varying the feed. Dynamic light scattering and transmission electron microscopy analysis indicated that PSPAESs self-assembled into spherical micelles in aqueous solutions. Interestingly, the micellar solution of PSPAESs prepared by dialysis was found to grow Cu2S nanowires on a Cu grid under ambient conditions. Formation of Cu2S nanowires on various substrates including a Si wafer and graphene was demonstrated in the presence of Cu and a sulfur source. UV-vis spectroscopy and X-ray photoelectron spectroscopy data suggests PSPAESs assist dissolution of metallic Cu into Cu(II) enabling the formation of Cu2S nanowires.
Evolution of supramolecular chirality from self-assembly of achiral compounds and control over its handedness is closely related to the evolution of life and development of supramolecular materials with desired handedness. Here we report a system where the entire process of induction, control and locking of supramolecular chirality can be manipulated by light. Combination of triphenylamine and diacetylene moieties in the molecular structure allows photoinduced self-assembly of the molecule into helical aggregates in a chlorinated solvent by visible light and covalent fixation of the aggregate via photopolymerization by ultraviolet light, respectively. By using visible circularly polarized light, the supramolecular chirality of the resulting aggregates is selectively and reversibly controlled by its rotational direction, and the desired supramolecular chirality can be arrested by irradiation with ultraviolet circularly polarized light. This methodology opens a route to ward the formation of supramolecular chiral conducting nanostructures from the self-assembly of achiral molecules.
Global production for NF3 is continuously increasing, especially due to its heavy consumption in the semiconductor industry. Even though the amount of its emission is relatively small compared to other greenhouse gases, particularly CO2, the relatively long atmospheric lifetime of NF3 makes its emission cumulative, possibly contributing to the global climate change. Membrane-based separation techniques are very promising for the energy-efficient NF3 recovery. It is, therefore, critically important to evaluate the N2/NF3 separation performance by using commercial polymeric membranes. Here, for the first time, the empirical N2/NF3 upper bound relationship is established by using a wide variety of commercial polymeric membranes including both glassy and rubbery polymers based on their single gas (i.e. N2 and NF3) permeation characterization. Among those tested, 6FDA–DAM:DABA (3:2), Teflon® AF 2400 and PTMSP exhibited relatively high N2/NF3 separation performance. The theoretical N2/NF3 upper bound curve was also defined and found comparable with our empirical upper bound limit. In an effort to improve the N2/NF3 separation performance, mixed matrix membranes were prepared by incorporating zeolitic imidazolate framework molecular sieves into Matrimid® 5218. The effects of solvents, particle sizes, and ligands on the transport properties in mixed matrix membranes were investigated.
We report synthesis of hierarchically porous polymers (HPPs) consisting of micropores and well-defined 3D continuous mesopores by combination of hyper-cross-linking and block polymer self-assembly. Copolymerization of 4-vinylbenzyl chloride (VBzCl) with divinylbenzene (DVB) in the presence of polylactide (PLA) macro-chain-transfer agent produced a cross-linked block polymer precursor PLA-b-P(VBzCl-co-DVB) via reversible addition–fragmentation chain transfer polymerization. A nanoscopic bicontinuous morphology containing PLA and P(VBzCl-co-DVB) microdomains was obtained as a result of polymerization-induced microphase separation. While a basic treatment of the precursor selectively removed PLA to yield a reticulated mesoporous polymer, hyper-cross-linking of the precursor by FeCl3 generated micropores in the P(VBzCl-co-DVB) microdomain via Friedel–Crafts alkylation and simultaneously degraded PLA to produce the HPP containing micropores in the mesoporous framework. The mesopore size of the HPP could be precisely controlled from 6 to 15 nm by controlling the molar mass of PLA. We demonstrate acceleration in adsorption rate in the HPP compared to a hyper-cross-linked microporous polymer.
Interfacial polymerization of an acid chloride-containing block polymer and a multivalent amine in the presence of a macroporous support was explored as a means to generate a nanoporous thin film composite (TFC) membrane potentially useful for ultrafiltration. When polylactide-b-poly(styrene-co-vinylbenzoyl chloride) (PLA-b-P(S-co-VBC)) in an organic phase and m-phenylenediamine (MPD) in an aqueous phase were used as the reactive block polymer and the amine, respectively, a block polymer thin film was successfully formed on a polysulfone support. This nanostructured film could be converted into a nanoporous layer by subsequent PLA etching under mild basic conditions. While most organic solvents used to dissolve PLA-b-P(S-co-VBC) damaged the support and decreased permeability of the resulting membrane, use of a mixture of methyl isobutyl ketone and acetonitrile produced a TFC membrane with high permeability.
Two donor-acceptor alternating copolymers based on electron-rich triarylamine, di(1-(6-(2-ethylhexyl))naphthyl)phenylamine (DNPA), and electron-deficient benzothiadiazole and benzoselenadiazole derivatives were designed and synthesized via Suzuki coupling reaction. The resulting triarylamine-based alternating copolymers PDNPADTBT and PDNPADTBS showed good solubility in common organic solvents and good thermal stability. The optical band gaps determined from the onset absorption were 1.93 and 1.81 eV, respectively. By introducing the naphthalene ring into the triarylamine, copolymers had relatively deep HOMO energy levels of −5.48 and −5.45 eV, which led to a high open circuit voltage (Voc) and good air stability for photovoltaic application. Bulk heterojunction solar cells were fabricated with a structure of ITO/PEDOT-PSS/copolymers-PC70BM/LiF/Al by blending the copolymer with PC70BM. Both blend systems showed remarkably high Voc near 0.9 V, and the highest performance of 2.2% was obtained from PDNPADTBT, with Voc = 0.88 V, Jsc = 7.4 mA/cm2, and a fill factor of 34.4% under AM 1.5 G.
Detailed experiments designed to optimize and understand the solvent vapor annealing of cylinder-forming poly(styrene)-block-poly(lactide) thin films for nanolithographic applications are reported. By combining climate-controlled solvent vapor annealing (including in situ probes of solvent concentration) with comparative small-angle X-ray scattering studies of solvent-swollen bulk polymers of identical composition, it is concluded that a narrow window of optimal solvent concentration occurs just on the ordered side of the order–disorder transition. In this window, the lateral correlation length of the hexagonally close-packed ordering, the defect density, and the cylinder orientation are simultaneously optimized, resulting in single-crystal-like ordering over 10 μm scales. The influences of polymer synthesis method, composition, molar mass, solvent vapor pressure, evaporation rate, and film thickness have all been assessed, confirming the generality of this behavior. Analogies to thermal annealing of elemental solids, in combination with an understanding of the effects of process parameters on annealing conditions, enable qualitative understanding of many of the key results and underscore the likely generality of the main conclusions. Pattern transfer via a Damascene-type approach verified the applicability for high-fidelity nanolithography, yielding large-area metal nanodot arrays with center-to-center spacing of 38 nm (diameter 19 nm). Finally, the predictive power of our findings was demonstrated by using small-angle X-ray scattering to predict optimal solvent annealing conditions for poly(styrene)-block-poly(lactide) films of low molar mass (18 kg mol–1). High-quality templates with cylinder center-to-center spacing of only 18 nm (diameter of 10 nm) were obtained. These comprehensive results have clear and important implications for optimization of pattern transfer templates and significantly advance the understanding of self-assembly in block copolymer thin films.
Controlled copolymerization of acryloyl chloride (AC), methacryloyl chloride (MAC), and vinylbenzoyl chloride (VBC) with styrene via the reversible addition–fragmentation chain transfer (RAFT) process was investigated. Copolymerization was conducted in 1,4-dioxane at 60 °C using azobisisobutyronitrile as an initiator and S-1-dodecyl-S′-(R,R′-dimethyl-R′′-acetic acid) trithiocarbonate as a chain transfer agent (CTA). The reactive copolymer was obtained by precipitating in hexanes. Methyl ester analogues of the reactive polymers were obtained by precipitating in methanol for analytical purposes and their 1H nuclear magnetic resonance spectroscopy and size exclusion chromatography analyses indicated that the best control was achieved for P(S-co-VBC) produced by copolymerization of styrene and VBC. Kinetics of the copolymerization of styrene and VBC was consistent with the RAFT mechanism. Reactive block polymers consisting of the P(S-co-VBC) block were also readily prepared using a macromolecular chain transfer agent. P(S-co-VBC) was successfully functionalized by reaction with alcohols or amines to form ester or amide linkages demonstrating its utility for the postpolymerization modification approach.
The viscosity of poly(styrene)-b-poly(lactide) [PS-b-PLA] solutions in a neutral solvent was characterized by magnetic microrheology. The effect of polymer concentration on the viscosity of the block polymer solutions was compared with that of the PS and PLA homopolymers in the same solvent. The viscosity of PS-b-PLA solution, unlike the homopolymer solutions, showed a steep increase over a narrow concentration range. The steep rise was concomitant with microphase separation into an ordered cylindrical microstructure as determined by small-angle X-ray scattering. Hence microrheology proved effective as a means of characterizing the order–disorder transition concentration. During an in situ drying experiment, changes in local viscosity through the depth of a block copolymer solution were characterized as a function of drying time. Early in the drying process, the viscosity rose steadily and was uniform through the depth, a result consistent with steadily increasing and uniform polymer concentration. However, later in the drying process as the overall polymer concentration approached that required for microphase separation, the viscosity of the polymer solution near the free surface became an order of magnitude higher than that near the bottom of the container. The zone of high viscosity moved downward as drying proceeded, consistent with a microphase separation front.
Using a simultaneous block polymerization/in situ cross-linking from a heterofunctional initiator approach, we produced a nanostructured and cross-linked block polymer in a single step from a ternary mixture of monomers and used it as a precursor for a cross-linked nanoporous material. Using 2-(benzylsulfanylthiocarbonylsulfanyl)ethanol as a heterofunctional initiator, simultaneous ring-opening transesterification polymerization of d,l-lactide in the presence of tin 2-ethylhexanoate as a catalyst and reversible addition–fragmentation chain transfer polymerization of styrene at 120 °C produced a polylactide-b-polystyrene (PLA-b-PS) block polymer. Incorporation of divinylbenzene in the polymerization mixture allowed in situ cross-linking during the simultaneous block polymerization to result in the cross-linked block polymer precursor in one step. This material was converted into cross-linked nanoporous polymer by etching PLA in a basic solution.












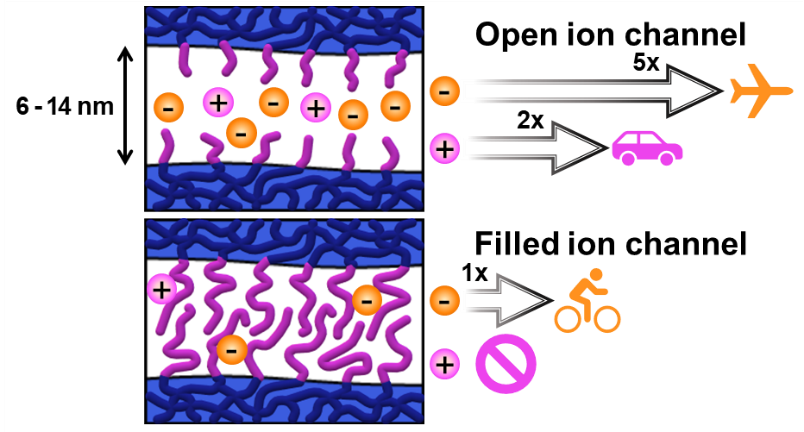


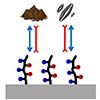



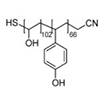
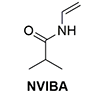
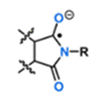




![[특집] 태초에 카이랄성은 어떻게 생겼을까: 빛에서 유래하는 비대칭성의 비밀](https://nanopsg.kaist.ac.kr/wp-content/uploads/2022/02/2022_JACS_thumb.jpg)

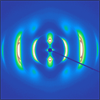
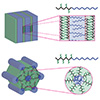



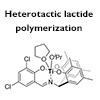




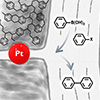




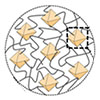



![[특집] 미세다공성 고분자의 세공 크기 제어 전략 (strategies for controlling pore size in microporous polymers)](https://nanopsg.kaist.ac.kr/wp-content/uploads/2020/12/2020_PST_thumb.jpg)


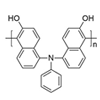
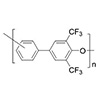


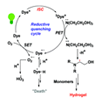



![[밝은빛 이용 우수연구논문] 중합에 의해 유도되는 미세상분리을 이용한 나노다공성 고분자 마이크로캡슐의 제조 연구 (fabrication of nanoporous polymer microcapsules by polymerization-induced microphase separation)](https://nanopsg.kaist.ac.kr/wp-content/uploads/2020/11/2018_ChemMater_thumb.jpg)










![[일반총설] 블록 공중합체 전구체로부터 유도되는 다공성 고분자 (porous polymers derived from block polymer precursors)](https://nanopsg.kaist.ac.kr/wp-content/uploads/2020/12/2015_PST_thumb.jpg)