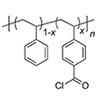
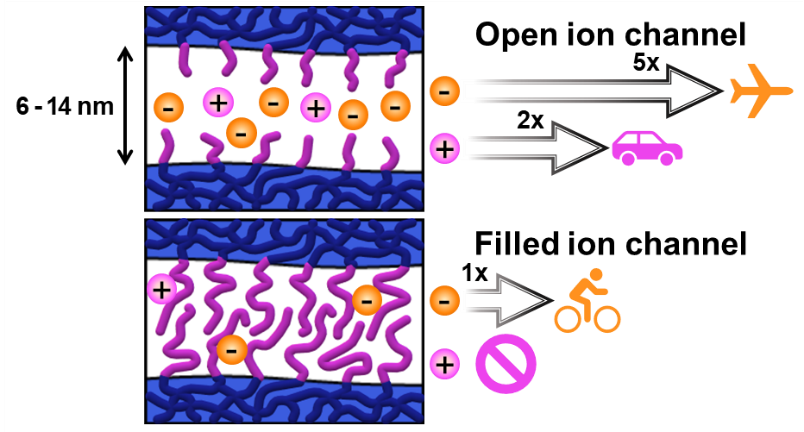
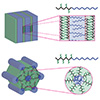
Permselective anion exchange membranes (AEMs) are of great interest in electrochemical devices, allowing the selective transport of species of interest through the membrane while preventing the crossover of undesired substrates. However, porous and nonporous membranes have traditionally been developed and applied separately for these applications. In this study, we utilized the merits of polymerization-induced microphase separation to observe the transition from porous to nonporous membranes by controlling the etchable block length and the hydrophilic domain size, respectively. At a certain point, a transition from porous to nonporous permeation behavior was observed, where permeation no longer depended on the open pore size. For the fabrication of AEMs with well-defined and 3D continuous nanochannels, we integrated an etchable sacrificial block with a pre-functional block containing precursors for quaternary ammonium groups to create a pore decorated with positively charged dangling chains. Etching and amination yielded the target AEM with a controlled pore size and ammonium ion density. Increasing the length of the dangling chains resulted in nonporous AEMs, which allowed exclusive permeation of a negatively charged dye over a positive one. This provides strong evidence that the solution-diffusion model applies to nonporous membranes. Tunable permselectivity over a wide range, combined with stability in organic solvents and alkaline conditions, suggests that this methodology holds significant potential for the development of membranes for advanced electrochemical systems.
Despite extensive studies on nanoporous membranes for regulating lithium-ion (Li⁺) flux in lithium (Li)-metal batteries, the pore size design has largely focused on very small (< 5 nm) or extremely large (> 20 nm) dimensions, overlooking the intermediate pore size range. This gap, particularly between 5 and 15 nm, has limited exploration of critical Li⁺ transport phenomena and their impact on improving cell performance. Here, we developed robust and free-standing polymeric films with three-dimensional (3D) continuous nanoporous channels, precisely tuned to pore diameters ranging from 5 to 14 nm and immobilized sulfonate groups. Our systematic investigations revealed how pore size and immobilized anionic groups correlated with Li⁺ conductivity and battery performance. Notably, sulfonate-functionalized channels promoted Li⁺ conductivity significantly within this optimal pore range compared to non-functionalized counterparts. In an ether-based electrolyte with 1 M lithium bis(fluorosulfonyl)imide (LiFSI), the Li⁺ conductivity peaked at a pore diameter of 10 nm. Furthermore, the mobility of Li⁺ was approximately 4.4 times faster than FSI⁻, resulting in reducing interfacial resistance and promoting uniform Li deposition. The sulfonated nanoporous membrane in Li|LiFePO₄ full cells with an N/P ratio of 2.3 delivered excellent cycling stability over 1000 cycles while retaining approximately 80 % of the initial capacity.
Accumulation of aquatic organisms on submerged sea vessel surfaces increases drag, resulting in higher fuel consumption and more greenhouse gas emissions. Polymer coatings hold promise to combat marine biofouling as more environmentally friendly alternatives to metal-based paints. While hydrophilic coatings are known to lose antifouling efficacy upon sediment adsorption, how to arrange hydrophobic segments and develop a sufficiently thick coating layer to suppress diatom deposition in the presence of silt and maximize the antifouling performance has been unknown. Here, we developed an effective and scalable antifouling coating by combining the polydopamine precoating method with well-defined amphiphilic copolymers synthesized using a controlled polymerization technique. Reversible addition-fragmentation chain transfer copolymerization of zwitterion-containing hydrophilic sulfobetaine methacrylate and hydrophobic trifluoroethyl methacrylate produced the target copolymers with control of composition, molecular weight, and sequence. Zr(IV)-mediated coordination bonds between polydopamine and sulfobetaine groups yielded >20 nm thick films stable for a month in seawater. The random copolymer sequence exposed both hydrophilic and hydrophobic groups on the outermost coating surface. Synergistic repelling diatom and silt led to the best antifouling performance at the optimal hydrophilic-hydrophobic balance. Superior antifouling efficacy was retained for a month and was also effective on stainless steel, suggesting the potential for practical application.
We report growing a polymer chain from the backbone of a bottlebrush polymer in the neat polymerization condition produces nanostructured polymer monoliths with ordered morphologies based on the Janus bottlebrush architecture. We installed a norbornene unit at the end of the polylactide macro-chain transfer agent (PLA-CTA) by single unit monomer insertion. We polymerized the resulting macromonomer via ring-opening metathesis polymerization to produce the PLA bottlebrush polymer, where a trithiocarbonate moiety remains on the backbone per every repeating unit. Neat polymerization of styrene in the presence of the PLA bottlebrush polymer proceeded in a grafting-from manner following the reversible addition–fragmentation chain transfer mechanism, resulting in a monolithic solid containing the doubly grafted PLA and polystyrene (PS) side chains. Polymerization-induced microphase separation (PIMS) spontaneously occurred, driven by the incompatibility between PLA and the growing PS segment. In contrast to the significant disordered fraction in PLA-b-PS produced with the linear PLA-CTA, the PLA/PS Janus bottlebrush polymer showed improved order across the investigated composition range. Formation of the asymmetric lamellae up to >80 vol % of PS indicated a strong preference for the lamellar symmetry of the Janus architecture. The in situ structured monoliths even exhibited narrower scattering peak widths compared to the solution-cast and annealed sample, suggesting the utility of the Janus PIMS process for facile preparation of ordered nanostructured materials with uniform domain size.
We investigated the bilayer-folded lamellar (Lf) mesophase appearing in the aqueous solution of amphiphilic random copolymers. A series of copolymers were synthesized by reversible addition–fragmentation chain transfer copolymerization of oligo(ethylene glycol) acrylate with alkyl acrylate with different alkyl chain lengths from octyl (C8) to octadecyl (C18). The alkyl acrylate composition was adjusted between 50–60 mol %. In the concentrated solution with the carbon number of the alkyl side chain higher than 10, the copolymers associated in water via hydrophobic interaction between the alkyl chains to produce micellar bilayers, which were periodically folded into bilayer-folded lamellae. The appearance of a small-angle X-ray scattering (SAXS) peak at a low scattering vector corresponding to >10 nm length scale clearly distinguished the bilayer-folded lamellae from the micellar lamellae with the domain spacing of 5–7 nm. Two-dimensional (2D) SAXS corroborated the presence of bilayer-folded lamellae developing perpendicularly to the micellar lamellae, which is consistent with our previous report. While the Lf phase was observed at room temperature for dodecyl (C12) and tetradecyl (C14) side chains that formed amorphous packing, crystalline hexadecyl (C16) and octadecyl (C18) chains seem to disturb bilayer folding. Heating the solution above the melting temperature of the alkyl chains produced the Lf phase with the largest folding height in the case of C16. The scaling relationship of the folding height to the carbon number supports the idea that the bending rigidity of the bilayer influences the length scale of folding.
We report the synthesis of the amphiphilic diblock copolymer, poly (vinyl phenol)-block-poly (vinyl alcohol), and its reversible formation of invertible core-shell nanoparticles. The diblock copolymer composed of poly (4-vinylphenol) (P4VPh) and poly (vinyl alcohol) (PVA) was prepared by the switchable reversible addition-fragmentation chain transfer polymerization (switchable-RAFT) of 4-acetoxystyrene and vinyl acetate and subsequent acetyl deprotection. The diblock copolymer was soluble in dimethyl sulfoxide (DMSO) but self-assembled into core-shell nanoparticles with the addition of H2O or tetrahydrofuran (THF). The formation and transition of the invertible core-shell system in response to solvent composition was characterized by 1H NMR, dynamic light scattering, and transmission electron microscopy. Interestingly, the fluorescent behavior of the P4VPh block was changed according to the morphological change of core-shell nanoparticle induced by solvent composition. With increasing the H2O content, the P4VPh chains in the DMSO solution collapsed and the diblock copolymers self-assembled to core-shell type micelles. Aggregation of P4VPh chains increased the local concentration of the phenolic group, resulting in fluorescence quenching. However, with the addition of THF to the DMSO solution, the inverted core-shell nanoparticles were formed where segregated P4VPh chains showed fluorescence.
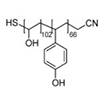
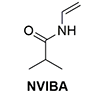
Thermo-responsive diblock copolymer, poly(N-isopropylacrylamide)-block-poly(N-vinylisobutyramide) was synthesized via switchable reversible addition–fragmentation chain transfer (RAFT) polymerization and its thermal transition behavior was studied. Poly(N-vinylisobutyramide) (PNVIBA), a structural isomer of poly(N-isopropylacrylamide) (PNIPAM) shows a thermo-response character but with a higher lower critical solution temperature (LCST) than PNIPAM. The chain extension of the PNVIBA block from the PNIPAM block proceeded in a controlled manner with a switchable chain transfer reagent, methyl 2-[methyl(4-pyridinyl)carbamothioylthio]propionate. In an aqueous solution, the diblock copolymer shows a thermo-responsive behavior but with a single LCST close to the LCST of PNVIBA, indicating that the interaction between the PNIPAM segment and the PNVIBA segment leads to cooperative aggregation during the self-assembly induced phase separation of the diblock copolymer in solution. Above the LCST of the PNIPAM block, the polymer chains begin to collapse, forming small aggregates, but further aggregation stumbled due to the PNVIBA segment of the diblock copolymer. However, as the temperature approached the LCST of the PNVIBA block, larger aggregates composed of clusters of small aggregates formed, resulting in an opaque solution.
Block polymers comprising covalently joined polymeric segments represent a class of nanostructured, multicomponent polymeric materials. Polymerization-induced microphase separation (PIMS) is an intriguing subset that allows for simultaneous nanostructuring during block polymer synthesis. In contrast to polymerization-induced self-assembly (PISA), useful for the spontaneous formation of block polymer micelles, PIMS is well suited to fabricating monolithic block polymer materials by turning a whole polymerization mixture into a nanostructured solid. With the in situ cross-linking feature, PIMS offers a facile route to nanostructured block polymer thermosets in combination with various polymerization techniques, from emulsion polymerization to 3D printing. This review aims to provide a comprehensive overview and practical guide on PIMS by covering its historical background and mechanistic aspects and also highlighting representative material classes and applicable polymerization techniques.
The present invention relates to a method of synthesizing hydrocarbon polymers using a deoxygenation reaction, wherein, by deoxygenating polymers including oxygen atom-containing functional groups in side chains thereof to thereby remove the functional groups of the side chains, various block copolymers including polyolefins and hydrocarbon polymers with complex architectures can be synthesized.
본 발명은 탈산소화반응을 이용한 탄화수소계 고분자의 합성방법에 관한 것으로서, 산소원자를 지니는 관능기를 측쇄에 포함하는 고분자의 탈산소화 반응을 이용해 측쇄의 관능기를 제거하여 폴리올레핀을 포함하는 다양한 블록 공중합체 및 복잡한 아키텍처의 탄화수소계 고분자를 합성할 수 있다.
Micrometer-sized aqueous droplets serve as a unique reactor that drives various chemical reactions not seen in bulk solutions. However, their utilization has been limited to the synthesis of low molecular weight products at low reactant concentrations (nM to μM). Moreover, the nature of chemical reactions occurring outside the droplet remains unknown. This study demonstrated that oil-confined aqueous microdroplets continuously generated hydroxyl radicals near the interface and enabled the synthesis of polymers at high reactant concentrations (mM to M), thus successfully converting the interfacial energy into the synthesis of polymeric materials. The polymerized products maintained the properties of controlled radical polymerization, and a triblock copolymer with tapered interfaces was prepared by the sequential addition of different monomers into the aqueous microdroplets. Furthermore, a polymerization reaction in the continuous oil phase was effectively achieved by the transport of the hydroxyl radicals through the oil/water interface. This interfacial phenomenon is also successfully applied to the chain extension of a hydrophilic polymer with an oil-soluble monomer across the microdroplet interface. Our comprehensive study of radical polymerization using compartmentalization in microdroplets is expected to have important implications for the emerging field of microdroplet chemistry and polymerization in cellular biochemistry without any invasive chemical initiators.
We report a synthetic methodology for decorating a surface of metal–organic frameworks (MOFs) with polymers through postsynthetic modification. Well-defined polymers with reversibly deactivated radical species at their chain end were reacted with vinyl-functionalized MOFs in the presence of a radical initiator. The radical addition forms a C–C bond between the polymer end with the functional group at the MOF ligand. We used sterically bulky star polymers containing electron-deficient maleimide chain ends, which facilitated modification of the external surface, yielding polymer-grafted MOF composite particles. A patchy MOF particle can also be obtained by simultaneously grafting two polymers and jammed at the immiscible liquid–liquid interface. We further show that the selective removal of a sacrificial polymer would partially expose the surface of MOFs to external environment, which hinders the uptake of macromolecular guests above the critical hydrodynamic size. Overall, four polymer@MOF composites have successfully been achieved through the present postsynthetic patchworks on MOFs with star polymers and selective etching process.
Randomness is perceived in two different extremes, in macroscopic homogeneity and local heterogeneity, but apparently far away from order. Here we show that a periodic order spontaneously arises from a binary random copolymer when self-assembly occurs in an ensemble containing > 1015 possible chain sequences. A Bernoullian distribution of hydrophilic and hydrophobic side chains grafted onto a linear backbone was constructed by random copolymerization. When the polymer chains associate in water, a sequence matching problem occurs because of the drastic heterogeneity in sequence: this is believed to generate local curvature mismatches which deviate from the ensemble-averaged interfacial curvature. Periodic folding of the self-assembled bilayer stabilizes the curvature instability as recurring hinges. Reminiscent of chain-folded lamellae found in polymer crystallization, this new liquid crystalline mesophase, characterized as bilayer-folded lamellae, manifests itself as an anisotropically alignable birefringent hydrogel with structural hierarchy across multiple length scales.
We report the synthesis and self-assembly of brush–linear diblock copolymers with variable side-chain length and density. Poly(pentafluorophenyl acrylate-g-ethylene glycol)-b-polystyrene ((PPFPA-g-PEG)-b-PS) brush–linear diblock copolymers are prepared by sequential reversible addition–fragmentation chain transfer (RAFT) polymerization of PPFPA and PS, followed by postpolymerization reaction between the precursor PPFPA-b-PS diblock copolymer and amine-functionalized PEG. By controlling the PEG chain length and the degree of substitution, we obtained brush–linear diblock copolymers with different side-chain lengths and densities. The solid-state morphologies of the diblocks are then examined by small-angle X-ray scattering (SAXS). At low PEG side-chain density, the segregation of PEG and PS away from PPFPA leads to the formation of PEG and PS lamellar domains with PPFPA in the interface. At high PEG side-chain density, the segregation is between the PPFPA-g-PEG brush block and the PS linear block, and the domain morphology is determined by the composition of the brush block. A partial experimental phase diagram is presented, and it illustrates the importance of both side-chain length and density on the microdomain morphology of brush–linear diblock copolymers.
Supramolecular polymerization offers a fascinating opportunity to develop dynamic soft materials by associating monomeric building blocks via noncovalent interactions. We report that polymerization can spontaneously drive the supramolecular polymerization of nanoscale micellar objects. We constructed the patchy micelles via two-step polymerization-induced self-assembly. A horizontal association between the patches results in a 1D supermicellar chain in situ by minimizing the enthalpic penalty of exposing the growing chains to solvent. Its length grows with increasing degree of polymerization, confirming that the supramolecular polymerization was triggered and controlled by polymerization. Our results highlight the observation that (1) the entire self-assembly process of forming, compartmentalizing, and associating the micelles can be driven by polymerization in a concerted manner and that (2) polymerization-induced self-assembly now can use compartmentalized nanoobjects as substrates beyond block copolymer chains. Polymerization-induced supramolecular polymerization could be useful for the autonomous preparation of hierarchical nanostructures.
The present invention relates to a method of preparing a hierarchically porous polymer and a hierarchically porous polymer prepared thereby. The method comprises the steps of: (a) polymerizing an external oil phase of a high internal phase emulsion (HIPE) consisting aqueous droplets to produce a cross-linked block copolymer; (b) obtaining a macroporous polymer with interconnected macropores by removing the aqueous droplets; and (c) treating the obtained porous polymer with a base, thereby obtaining a hierarchically porous polymer having three-dimensional mesopores formed in the macroporous walls. According to the method, the macropore size and mesopore size of the hierarchically porous polymer can all be controlled. The hierarchically porous polymer prepared by the method can easily separate polymers having different sizes, and thus is highly useful in the polymer separation field.
We report the synthesis of thiopropionic acid-functionalized polymethacrylate-b-polystyrene (PMATA-b-PS) as a template for patterning Ag nanoarrays. Reversible addition-fragmentation chain transfer (RAFT) polymerization of silyl-protected propagyl methacrylate followed by chain extension with styrene produced a block copolymer precursor. Deprotection of the trimethylsilyl group and subsequent thiol-yne reaction afforded the target, PMATA-b-PS. While the entire series of the precursor was in the disordered phase, microphase separated morphologies were identified from PMATA-b-PS, indicating an increased interaction parameter (χ) as a result of introducing the acid groups. As a preliminary result, we show that an Ag nanoparticle array can be fabricated by selectively associating Ag+ ions to the PMATA cylinders in the thin film of PMATA-b-PS and removing the organic polymer layer by oxygen plasma treatment.
This study explores hyper-cross-linking of the cores of block copolymer micelles as a means to generate star polymers with a hyper-cross-linked core surrounded by linear corona arms. A solution of poly(methyl methacrylate)-b-polystyrene (MS) was prepared in acetonitrile to form micelles which were reacted with α,α’-dichloro-p-xylene in the presence of FeCl3 to produce core hyper-cross-linked star (CHS) polymers by selective cross-linking of the PS core. A kinetic investigation showed formation of high-molar mass species (>104 kg mol−1) within 1 h of reaction, which supported conversion of individual MS micelles into CHS polymers. We synthesized several CHS polymers by varying the PS core fractions from 20 to 53%. All the polymers possessed discrete spherical cores that were 19–60 nm in diameter and all were highly soluble in organic solvents retaining the CHS architecture. While permanent microporosity was not detected by gas sorption measurements, increased dye uptake of CHS polymer in solution suggests utility of CHS polymers as stable and solution-processible nanocontainers with accessible free volume in the core.
We report polymerization-induced self-assembly via controlled cross-linking copolymerization to produce robust block copolymer micelles with spherical, elongated, and branched shapes. Reversible addition–fragmentation chain transfer (RAFT) copolymerization of styrene and divinylbenzene (DVB) or 1,2-bismaleimidoethane (BMI) as a cross-linker in the presence of a polylactide macro-chain transfer agent (PLA-CTA) was performed in acetonitrile, which is a non-solvent to polystyrene (PS). The addition of the cross-linker accelerates the copolymerization compared to styrene homopolymerization, which leads to the formation of block polymer micelles within a shorter time frame, followed by in situ inter-chain cross-linking. The micelles are virtually identical to the core cross-linked star polymer consisting of a cross-linked polystyrenic core surrounded by a PLA corona. Molecular weights up to more than 1000 kg mol−1 could be obtained with relatively narrow dispersity values (1.1–1.4). In the case of copolymerization with DVB, the micellar morphology changes from spherical to elongated and branched shapes with increasing conversion. The size and morphology of the micelles are retained in a good solvent to PS, suggesting that the in situ cross-linking effectively stabilizes the micellar core. BMI undergoes alternating copolymerization with styrene in the early stage of polymerization and yields spherical micelles exclusively, because the densely cross-linked core seems to prevent further morphological transition.
Mesoporous nonoxide ceramics are attractive for applications such as catalytic supporters and separations with exceptional thermochemical stability. Here we report on the one-step preparation of microphase-separated bicontinuous organic–inorganic polymer precursors for forming 3D continuous polymer-derived ceramic monoliths without an external block copolymer template and annealing steps. We combined polymerization-induced phase separation with in situ hybrid block polymer formation from a mixture of a preceramic monomer, a cross-linker, and a thermally decomposable organic segment containing a terminal chain transfer agent. The resultant cross-linked polymeric monoliths, moldable to any desired shape, were converted to 3D-continuous mesoporous silicon carbonitride ceramics with a pore size in the 3–11 nm range and a surface area of 107–410 m2 g–1 by varying the molar mass of the sacrificial organic block and the pyrolytic temperature. The 3D-disordered pore structure is beneficial for retaining the monolithic shape via isotropic shrinkage during ceramization. The distinctive characteristics of this synthetic approach, which are the absence of a solvent, a structure-directing block copolymer, and an annealing process, are affordable for the large production of nanoporous ceramic monoliths for various high-temperature applications and should be applicable for additive manufacturing with direct polymerizability for the fabrication of hierarchically porous materials in complex shapes with dimensional scalability.
We developed a block polymer-based synthetic route to sulfonated porous composites (SPCs) with precisely controlled nanopore size. By reducing the pore size to <4 nm and introducing a high density of surface sulfonic acid, the permeation of vanadium ions was effectively suppressed. We employed a polymerization-induced microphase separation (PIMS) process, in which a polyethylene fiber mat impregnated with a liquid polymerization mixture was spontaneously transformed into a fiber-reinforced and cross-linked block polymer membrane. Selective etching and sulfonation then produced the target composite membrane. In a vanadium redox flow battery (VRFB) cell, an SPC with 3.6 nm-sized mesopores, 109 m2 g–1 of specific surface area, and 0.3 mL g–1 of mesoporosity outperformed a Nafion 212 membrane of similar thickness, providing higher proton conductivity and much lower vanadium permeability. Thanks to the composite reinforcement, the membrane demonstrated remarkably enhanced mechanical stability. The SPC membrane could be successfully operated up to 300 cycles. Compared with Nafion 212, the SPC exhibited higher energy efficiencies (EEs) and higher discharge capacity retention. These results suggest the promise of block polymer-based permselective membranes in advanced battery applications.
We report the synthesis of a series of statistical terpolymer poly[(methyl methacrylate)-co-lauryl methacrylate-co-2-((3,5-bis(4-carbamoyl-3-(trifluoromethyl)phenoxy)benzyloxy)carbonylamino)ethyl methacrylate] (P(MMA-co-LMA-co-BMA)) by reversible addition–fragmentation chain transfer polymerization and their aggregation behaviors in solution. In toluene, the solution behavior of terpolymer was controlled by the molar fractions of lauryl methacrylate (LMA) and benzamide-containing methacrylate (BMA) in the polymer, which increased solubility and promoted hydrogen bonding between the primary aromatic amides, respectively. Temperature-dependent 1H NMR spectroscopy also indicated gradual dissociation of the hydrogen bonds with increasing temperature. For the polymer containing 2.7 mol % of LMA and 2.7 mol % of BMA repeating units, we demonstrated that dissolving the polymer in tetrahydrofuran as a good solvent and switching the solvent with toluene produced polymer nanoparticles with diameters of several tens of nanometers, as observed by dynamic light scattering. Intramolecular hydrogen bonding was dominant and induced the noncovalent chain collapse. When the temperature of the particle dispersion in toluene at a concentration > 30 mg/mL was increased from RT to 50 °C, a significant increase in viscosity was observed. This behavior was not observed in a toluene solution of poly(methyl methacrylate), which showed decreased viscosity at a higher temperature. The viscosity increase was accompanied by a decrease in the particle size, and both were attributed to the dissociation of some intramolecular hydrogen bonds within the particles, which can increase the number of individual chains in toluene and result in more intermolecular interactions.
We report the self-assembly of monolayer vesicles from Janus core–shell bottlebrush polymers. A route was developed to synthesize doubly grafted bottlebrush copolymers (DGBCPs) possessing A-b-B and B′-b-C side chains on a single repeating unit. Graft-through ring-opening metathesis polymerization of a norbornene moiety installed by single unit monomer insertion allowed us to place the backbone on any repeating unit of the core (B and B′) block. By decorating each core chain end with different chains via reversible addition–fragmentation chain transfer polymerization, we can obtain nanoobjects with an asymmetric B core and a phase-separated A/C shell. We demonstrate that polystyrene-branch-polystyrene′ and polylactide-b-polystyrene-branch-polystyrene′-b-poly(n-butyl acrylate) macromonomers can be successfully synthesized and polymerized to produce DGBCPs in high yields (81–94% conversion) with an absolute molar mass of 149–395 kg mol–1 and a dispersity of 1.18–1.38. In a solvent slightly more selective to A than C, self-assembly of monolayer vesicles with diameter of <100 nm was observed by transmission electron microscopy. Dissipative particle dynamics simulations suggest that increasing the backbone length and moving the backbone toward the B′/C interface increases the backbone bending energy and favors a lower curvature. The spontaneous curvature appears to prefer a particular layer radius, avoiding bilayer formation.
Heteroarm core cross-linked star (CCS) polymers consist of two different polymer chains covalently joined to a cross-linked core. We investigated their self-assembly behavior to understand whether intramolecular segregation can be induced during synthesis, to produce spatial domains enriched with each polymer, and whether they would exhibit well-defined microphase separation morphologies as a result. Heteroarm CCS polymers containing polylactide (PLA) and polystyrene (PS) arms were synthesized by reversible addition–fragmentation chain transfer copolymerization of styrene and 1,2-bis(maleimidoethane) in the presence of a PLA-macro chain transfer agent (PLA-CTA), followed by chain extension with styrene (the in–out route). Dynamic light scattering, transmission electron microscopy, and small angle X-ray scattering analyses were employed to examine the self-assembly behavior in toluene and acetonitrile, as a relatively neutral and a PLA-selective solvent, respectively. Above a critical PS molar mass, lamellar-like and spherical morphologies were observed, formed by microphase separation into discrete PLA and PS domains. The increase in order with increasing PS molar mass was consistent with the segregation strength-dependent microphase separation behavior. In contrast, when the CCS polymer was synthesized by simultaneously joining PLA and PS chains (the multi macroinitiatior route) it produced rather ill-defined self-assemblies, suggesting that styrene chain extension via the in–out process is important to achieve intramolecular segregation. Using the more PLA-selective acetonitrile as a polymerization solvent indeed produced more well-defined supermicelles with PS cores and PLA coronas, confirming that intramolecular segregation can be driven by the incompatibility of the growing PS to the intramolecular environment, including PLA and the solvent.
We propose the defunctionalization of vinyl polymers as a strategy to access previously inaccessible polyolefin materials. By utilizing B(C6F5)3-catalyzed deoxygenation in the presence of silane, we demonstrate that eliminating the pendent ester in poly(methyl acrylate) effectively leaves a linear hydrocarbon polymer with methyl pendants, which is polypropylene. We further show that a polypropylene-b-polystyrene diblock copolymer and a polystyrene-b-polypropylene-b-polystyrene triblock copolymer can be successfully derived from the poly(methyl acrylate)-containing block polymer precursors and exhibit quite distinct materials properties due to their chemical transformation. This unique postpolymerization modification methodology, which goes beyond the typical functional group conversion, can offer access to a diverse range of unprecedented polyolefin block polymers with a variable degree of functional groups.
We report self-assembly of a statistical copolymer poly(styrene-co-methyl methacrylate) (P(S-co-MMA)) containing ionic liquid (IL)-philic methyl methacrylate (MMA) and IL-phobic styrene (S) repeating units in IL for fabrication of electrolyte-gated organic transistors. P(S-co-MMA)s with high MMA contents were synthesized by copolymerization of styrene and MMA via a reversible addition–fragmentation chain transfer (RAFT) process, and their behavior in 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([EMI][TFSI]) was investigated. While dynamic light scattering analysis showed formation of a micellar solution at low concentration, the elastic modulus of the viscoelastic solution increased significantly more than the loss modulus at high concentration. Small angle X-ray scattering analysis suggested ill-defined phase separation between PS-rich segments and PS-lean segments swollen in [EMI][TFSI]. The resulting P(S-co-MMA)/[EMI][TFSI] mixture exhibited increased ionic conductivity compared to the PS-b-PMMA-b-PS block polymer gel, as well as superior device performance in transistor gating experiments.
We report the synthesis of microporous hyper-cross-linked polymers (HCPs) with increased specific surface area and porosity by the in situ removal of trimethylsilyl (TMS) groups during hyper-cross-linking. We synthesized poly(4-trimethylsilylstyrene-co-vinylbenzyl chloride-co-divinylbenzene)s (P(TMSS-co-VBzCl-co-DVB)s) with different compositions by reversible addition–fragmentation chain transfer copolymerization and converted them into HCPs by reacting with FeCl3 in 1,2-dichloroethane. The nearly quantitative removal of the TMS groups was observed during the reaction following the electrophilic aromatic substitution mechanism, where the TMS group shows higher reactivity than an aromatic hydrogen. Substantial enhancement in pore characteristics including surface area, microporosity, and mesoporosity was noticed up to a certain level of TMSS incorporation, compared with HCP derived from P(VBzCl-co-DVB). We suggest the porogenic TMS group increases porosity mainly by in situ removal via facilitated substitution reaction, which creates permanent voids in the hyper-cross-linked network. The use of TMSS provides a feasible and complementary route to tuning the pore characteristics of HCPs by varying DVB content, and is applicable to the synthesis of hierarchically porous polymers containing micropores within a mesoporous framework from block polymer precursors.
We investigated proton conductivity and the permeability of monovalent cations across sulfonated mesoporous membranes (SMMs) prepared with well-defined pore sizes and adjustable sulfonic acid content. Mesoporous membranes with three-dimensionally continuous pore structure were produced by the polymerization-induced microphase separation (PIMS) process involving the reversible addition–fragmentation chain transfer (RAFT) copolymerization of styrene and divinylbenzene in the presence of a polylactide (PLA) macrochain transfer agent and subsequent PLA etching. This allowed us to control pore size by varying PLA molar mass. Postsulfonation of the mesoporous membranes yielded SMMs whose pore structure was retained. The sulfonic acid content was adjusted by reaction time. While proton conductivity increased with increasing ion exchange capacity (IEC) without noticeable dependence on the pore size, ion permeability was strongly influenced by the pore size and IEC values. Decreasing pore size and increasing IEC resulted in a decrease in ion permeability, suggesting that ions traverse across the membrane via the vehicular mechanism, through the mesoporous spaces filled with water. We further observed that the permeability of the vanadium oxide ion was dramatically suppressed by reducing the pore size below 4 nm, which was consistent with preliminary vanadium redox flow battery data. Our approach suggests a route to developing permselective membranes by decoupling proton conductivity and ion permeability, which could be useful for designing separator materials for next-generation battery systems.
We report a blending mechanism of polystyrene-b-poly(ethylene oxide) (PS-b-PEO) and PS homopolymer (homoPS) at the air/water interface. Our blending mechanism is completely different from the well-known “wet–dry brush theory” for bulk blends; regardless of the size of homoPS, the domain size increased and the morphology changed without macrophase separation, whereas the homoPS of small molecular weight (MW) leads to a transition after blending into the block copolymer domains, and the large MW homoPS is phase-separated in bulk. The difference in blending mechanism at the interface is attributed to adsorption kinetics at a water/spreading solvent interface. Upon spreading, PS-b-PEO is rapidly adsorbed to the water/spreading solvent interface and forms domain first, and then homoPS accumulates on them as the solvent completely evaporates. On the basis of our proposed mechanism, we demonstrate that rapid PS-b-PEO adsorption is crucial to determine the final morphology of the blends. We additionally found that spreading preformed self-assemblies of the blends slowed down the adsorption, causing them to behave similar to bulk blends, following the “wet–dry brush theory”. This new mechanism provides useful information for various block copolymer-homopolymer blending systems with large fluid/fluid interfaces such as emulsions and foams.
We report the preparation of hierarchically porous polymers containing fully interconnected and controlled micro-, meso-, and macropores, where a hyper-cross-linked microporous polymer skeleton forms a reticulating mesoporous wall that supports a highly porous macropore framework. These materials provide high specific surface area and >90% porosity, useful for rapid sorption of organic molecules.
Recently, because of rapid advances in electrical vehicles, unmanned air vehicles, and humanoid mobile robots, structural energy storage devices with a concurrent capability to store electrochemical energy and to support mechanical loads have been in the spotlight. However, a big hurdle to realizing an integrated electro-chemo-mechanical system is to develop highly compatible active electrodes and structural electrolytes with superior mechanical strength and electrochemical functionality while retaining light weight. We report a load-bearing structural supercapacitor by utilizing a bicontinuous PEO-b-P(S-co-DVB) structural electrolyte and carbon-coated Ni-Co nanowires grown on carbon fiber woven fabric. A liquid polymerization mixture between the electrodes is transformed into a solid-state block copolymer electrolyte, preserving conformal contact with the nanostructured electrode surface. The polymerization-induced microphase separation produces a bicontinuous morphology of cross-linked hard domain and liquid-like conductive domain in the electrode, providing high modulus and high conductivity. The resulting structural supercapacitor is able to operate under tensile and even bending load, suggesting its wide potential applications.
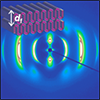
Microcapsules with nanoporous membranes can regulate transmembrane transport in a size-dependent fashion while protecting active materials in the core from the surrounding, and are thereby useful as artificial cell models, carriers for cells and catalysts, and microsensors. In this work, we report a pragmatic microfluidic approach to producing such semipermeable microcapsules with precise control of the cutoff threshold of permeation. Using a homogeneous polymerization mixture for the polymerization-induced microphase separation (PIMS) process as the oil phase of water-in-oil-in-water (W/O/W) double emulsions, a densely cross-linked shell composed of a bicontinuous nanostructure that percolates through the entire thickness is prepared, which serves as a template for a monolithic nanoporous membrane of microcapsules with size-selective permeability. We demonstrate that the nanopores with precisely controlled size by the block polymer self-assembly govern molecular diffusion through the membrane and render manipulation of the cutoff threshold.
We report on the phase separation behaviors of polymerization mixtures containing a polylactide macro-chain transfer agent (PLA-CTA), styrene, divinylbenzene, hydroxyl-terminated PLA (PLA-OH), and a molecular chain transfer agent which enable the ability to tune the pore size of a cross-linked polymer monolith in a facile manner. Cross-linked monoliths were produced from the mixtures via reversible addition-fragmentation chain transfer (RAFT) polymerization and converted into cross-linked porous polymers by selective removal of PLA while retaining the parent morphology. We demonstrate that pore sizes are tunable over a wide range of length scales from the meso- to macroporous regimes by adjusting the ratio of PLA-CTA to PLA-OH in the reaction mixture which causes the phase separation mechanism to change from polymerization-induced microphase separation to polymerization-induced phase separation. The possibility of increasing porosity and inducing simultaneous micro- and macrophase separation was also realized by adjustments in the molar mass of PLA which enabled the synthesis of hierarchically meso- and macroporous polymers.
We explored reversible addition-fragmentation chain transfer (RAFT) copolymerization of 1,2-bis(maleimidoethane) (BMI) with styrene (S) in the presence of polylactide macro-chain transfer agent (PLA-CTA) as a means to synthesize heteroarm core cross-linked star (CCS) polymers consisting of PLA and PS arms (PLAnPSn). Because of the strong alternating tendency of maleimide and styrenic double bonds, copolymerization of BMI with an excess of S depleted BMI in the early stage of polymerization forming a cross-linked core. The remaining S was successively polymerized to grow PS arms from the core, completing PLAnPSn via “in–out” mechanism. Use of a stoichiometric amount of S produced PLAn, which could be used as a macro-CTA for the synthesis of more well-defined PLAnPSn. Compared with divinylbenzene, copolymerization of BMI with S was much more effective for core formation suggesting the importance of the alternating character of the copolymerization. While PLAnPSn existed as stable nanoparticles in a neutral solvent in contrast to linear PLA-b-PS, it also self-assembled to form microphase-separated structures in a selective solvent and in bulk indicating that PLA and PS arms can be intramolecularly segregated.
We report on the use of photoinitiated reversible addition–fragmentation chain transfer (RAFT) polymerization for the facile fabrication of cross-linked nanoporous polymer films with three-dimensionally (3D) continuous pore structure. The photoinitiated polymerization of isobornyl acrylate (IBA) in the presence of 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (CTA) and 2,2-dimethoxy-2-phenylacetophenone as a photoinitiator proceeded in a controlled manner, yet more rapidly compared to thermally initiated polymerization. When polylactide-macroCTA (PLA-CTA) was used, PLA-b-PIBA with high molar mass was obtained after several minutes of irradiation at room temperature. We confirmed that microphase separation occurs in the PLA-b-PIBA and that nanoporous PIBA can be derived from the PLA-b-PIBA precursor by selective PLA etching. To fabricate the cross-linked nanoporous polymer, IBA was copolymerized with ethylene glycol diacrylate (EGDA) in the presence of PLA-CTA to produce a cross-linked block polymer precursor consisting of bicontinuous PLA and P(IBA-co-EGDA) microdomains, via polymerization-induced microphase separation. We demonstrated that nanoporous P(IBA-co-EGDA) monoliths and films with 3D continuous pores can be readily obtained via this approach.
Interfacial polymerization of an acid chloride-containing block polymer and a multivalent amine in the presence of a macroporous support was explored as a means to generate a nanoporous thin film composite (TFC) membrane potentially useful for ultrafiltration. When polylactide-b-poly(styrene-co-vinylbenzoyl chloride) (PLA-b-P(S-co-VBC)) in an organic phase and m-phenylenediamine (MPD) in an aqueous phase were used as the reactive block polymer and the amine, respectively, a block polymer thin film was successfully formed on a polysulfone support. This nanostructured film could be converted into a nanoporous layer by subsequent PLA etching under mild basic conditions. While most organic solvents used to dissolve PLA-b-P(S-co-VBC) damaged the support and decreased permeability of the resulting membrane, use of a mixture of methyl isobutyl ketone and acetonitrile produced a TFC membrane with high permeability.
Detailed experiments designed to optimize and understand the solvent vapor annealing of cylinder-forming poly(styrene)-block-poly(lactide) thin films for nanolithographic applications are reported. By combining climate-controlled solvent vapor annealing (including in situ probes of solvent concentration) with comparative small-angle X-ray scattering studies of solvent-swollen bulk polymers of identical composition, it is concluded that a narrow window of optimal solvent concentration occurs just on the ordered side of the order–disorder transition. In this window, the lateral correlation length of the hexagonally close-packed ordering, the defect density, and the cylinder orientation are simultaneously optimized, resulting in single-crystal-like ordering over 10 μm scales. The influences of polymer synthesis method, composition, molar mass, solvent vapor pressure, evaporation rate, and film thickness have all been assessed, confirming the generality of this behavior. Analogies to thermal annealing of elemental solids, in combination with an understanding of the effects of process parameters on annealing conditions, enable qualitative understanding of many of the key results and underscore the likely generality of the main conclusions. Pattern transfer via a Damascene-type approach verified the applicability for high-fidelity nanolithography, yielding large-area metal nanodot arrays with center-to-center spacing of 38 nm (diameter 19 nm). Finally, the predictive power of our findings was demonstrated by using small-angle X-ray scattering to predict optimal solvent annealing conditions for poly(styrene)-block-poly(lactide) films of low molar mass (18 kg mol–1). High-quality templates with cylinder center-to-center spacing of only 18 nm (diameter of 10 nm) were obtained. These comprehensive results have clear and important implications for optimization of pattern transfer templates and significantly advance the understanding of self-assembly in block copolymer thin films.
Controlled copolymerization of acryloyl chloride (AC), methacryloyl chloride (MAC), and vinylbenzoyl chloride (VBC) with styrene via the reversible addition–fragmentation chain transfer (RAFT) process was investigated. Copolymerization was conducted in 1,4-dioxane at 60 °C using azobisisobutyronitrile as an initiator and S-1-dodecyl-S′-(R,R′-dimethyl-R′′-acetic acid) trithiocarbonate as a chain transfer agent (CTA). The reactive copolymer was obtained by precipitating in hexanes. Methyl ester analogues of the reactive polymers were obtained by precipitating in methanol for analytical purposes and their 1H nuclear magnetic resonance spectroscopy and size exclusion chromatography analyses indicated that the best control was achieved for P(S-co-VBC) produced by copolymerization of styrene and VBC. Kinetics of the copolymerization of styrene and VBC was consistent with the RAFT mechanism. Reactive block polymers consisting of the P(S-co-VBC) block were also readily prepared using a macromolecular chain transfer agent. P(S-co-VBC) was successfully functionalized by reaction with alcohols or amines to form ester or amide linkages demonstrating its utility for the postpolymerization modification approach.
Using a simultaneous block polymerization/in situ cross-linking from a heterofunctional initiator approach, we produced a nanostructured and cross-linked block polymer in a single step from a ternary mixture of monomers and used it as a precursor for a cross-linked nanoporous material. Using 2-(benzylsulfanylthiocarbonylsulfanyl)ethanol as a heterofunctional initiator, simultaneous ring-opening transesterification polymerization of d,l-lactide in the presence of tin 2-ethylhexanoate as a catalyst and reversible addition–fragmentation chain transfer polymerization of styrene at 120 °C produced a polylactide-b-polystyrene (PLA-b-PS) block polymer. Incorporation of divinylbenzene in the polymerization mixture allowed in situ cross-linking during the simultaneous block polymerization to result in the cross-linked block polymer precursor in one step. This material was converted into cross-linked nanoporous polymer by etching PLA in a basic solution.

























![[밝은빛 이용 우수연구논문] 중합에 의해 유도되는 미세상분리을 이용한 나노다공성 고분자 마이크로캡슐의 제조 연구 (fabrication of nanoporous polymer microcapsules by polymerization-induced microphase separation)](https://nanopsg.kaist.ac.kr/wp-content/uploads/2020/11/2018_ChemMater_thumb.jpg)