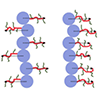
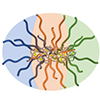
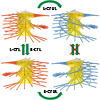
Multicompartment polymer nanoparticles, such as two-faced Janus and patchy particles composed of distinct chemical features, have received increasing attention because of their utility for interfacial and self-assembly applications originating from the asymmetric particulate structure. This review discusses such nanoparticles in several tens of nanometers produced by controlled polymerizations, which enable scalable synthesis with control of molecular characteristics. We focus on miktoarm core cross-linked star polymers and Janus core–shell bottlebrush polymers as 0D and 1D anisotropic nano-objects containing a discrete core and a compartmentalized shell. We discuss how controlled polymerizations can covalently build such complex architectures with spatial control of the constituting segments to achieve intramolecular segregation. Then, we collectively view their distinct interfacial and self-assembling behaviors reported in the literature from experimental and simulation perspectives.
Polymer grafting density critically influences the self-assembly of polymer-grafted nanoparticles, yet the low grafting density regime remains underexplored. Here, we investigate the thin-film self-assembly of bottlebrush polymer-grafted core/shell nanoparticles (BPGNPs) under quasi-2D confinement at near-zero grafting densities through coarse-grained molecular dynamics (CGMD). The NP core is modeled using a hard-core/soft-shoulder (HCSS) potential, and it is compared against Weeks–Chandler–Andersen (WCA) potential. While the phase behaviors of both models are well-known, the distinct phase behaviors of both models persist even with polymer grafting offering additional room for tunability. Unlike sufficiently high grafting density or bare nanoparticles (NPs), grafting a single bottlebrush polymer breaks the rotational symmetry. The resulting structural polarity of grafted NPs can be precisely controlled through bottlebrush design parameters. We demonstrate that enhanced structural polarity stabilizes specific ordered phases, enabling precise control over self-assembled morphologies such as hexagonal lattices, square lattices, and linear clusters. Lastly, we explore the impact of synthesis-induced heterogeneity by introducing bare NPs, dual-polymer-grafted particles, and unconjugated polymers as minor species, providing insights into morphological stability under realistic grafting conditions. This work advances our understanding of BPGNP self-assembly in the near-zero grafting density regime and establishes design principles for functional nanotechnology applications.
Nature utilizes self-assembly to form complex, functional structures, inspiring advanced materials design. Polymer crystallization drives assemblies with both ordered and disordered regions. Crystallization-driven assembly of BCPs enables unique hierarchical nanostructures with enhanced colloidal stability and directionality, applicable from optoelectronics to biomedicine. However, mechanisms governing morphological transitions remain poorly understood due to complex microphase separation and competitive crystallization. Using liquid-phase transmission electron microscopy, we visualize the spontaneous assembly of semicrystalline amphiphilic BCPs. We observe structural transformations from unimers to spherical, cylindrical, toroidal micelles, and vesicles by varying constituent block ratios. Image segmentation overcomes low contrast of aqueous assemblies, enabling motion tracking. Nanostructures exhibit structural evolution driven by long-range hydrophobic interactions from formed elemental micelles undergoing anomalous diffusion. Notably, toroid formation follows a distinct pathway compared with conventional BCPs due to semicrystalline BCPs’ preference for low curvature at the core-corona interface. Insights into assembly dynamics via real-time imaging provide strategies for controlling complex hierarchical structures.
Macromolecular self-assembly is essential in life and interfacial science. A macromolecule consisting of chemically distinct components tends to self-assemble in a selective solvent to minimize the exposure of the solvophobic segments to the medium while the solvophilic segments adopt extended conformations. While micelles composed of linear block copolymers represent classic examples of such solution assembly, recent interest focuses on the self-assembly of complex macromolecules with nonlinear architectures, such as star, graft, and bottlebrush. Such macromolecules include several to hundreds of polymer chains covalently tied to a core and a backbone. The pre-programmed, non-exchangeable chain arrangement makes a huge difference in their self-assembly. The field has witnessed tremendous advances in synthetic methodologies to construct the desired architectures, leading to discoveries of exotic self-assembly behavior. Thanks to the rapid evolution of computing power, computer simulation has also been an emerging and complementary approach for understanding the association mechanism and further predicting the self-assembling morphologies. However, simulating the self-assembly of architected macromolecules has posed a challenge as a huge number of objects should be included in the simulations. Comparing experimental results with simulations is not always straightforward, as synthetic routes to well-defined model systems with systematically controlled structural parameters are not often available. In this manuscript, we propose to bridge a gap between experiments and simulations in self-assembly of architected macromolecules. We focus on the key articles in this area reporting experimental evidence and simulation details and also cover recent examples in the literature. We start with discussing simulation methodologies applicable to investigate solution self-assembly across multiple levels of chemical resolution from all-atom to particle dynamics. Then, we delve into topological design, synthesis, and simulation of nonlinear macromolecules, including dendritic/star, network, and graft/bottlebrush polymers, to understand the architectural effect on the self-assembly behavior. We expand our discourse to embrace recent advances toward realizing more complex systems. For example, self-assembly in the presence of strong Coulombic interactions, such as in the case of polyelectrolytes, geometric constraints, and other components in solutions, exemplified by inorganic fillers, are introduced. Finally, the challenges and perspectives are discussed in the final section of the manuscript.
We investigated the bilayer-folded lamellar (Lf) mesophase appearing in the aqueous solution of amphiphilic random copolymers. A series of copolymers were synthesized by reversible addition–fragmentation chain transfer copolymerization of oligo(ethylene glycol) acrylate with alkyl acrylate with different alkyl chain lengths from octyl (C8) to octadecyl (C18). The alkyl acrylate composition was adjusted between 50–60 mol %. In the concentrated solution with the carbon number of the alkyl side chain higher than 10, the copolymers associated in water via hydrophobic interaction between the alkyl chains to produce micellar bilayers, which were periodically folded into bilayer-folded lamellae. The appearance of a small-angle X-ray scattering (SAXS) peak at a low scattering vector corresponding to >10 nm length scale clearly distinguished the bilayer-folded lamellae from the micellar lamellae with the domain spacing of 5–7 nm. Two-dimensional (2D) SAXS corroborated the presence of bilayer-folded lamellae developing perpendicularly to the micellar lamellae, which is consistent with our previous report. While the Lf phase was observed at room temperature for dodecyl (C12) and tetradecyl (C14) side chains that formed amorphous packing, crystalline hexadecyl (C16) and octadecyl (C18) chains seem to disturb bilayer folding. Heating the solution above the melting temperature of the alkyl chains produced the Lf phase with the largest folding height in the case of C16. The scaling relationship of the folding height to the carbon number supports the idea that the bending rigidity of the bilayer influences the length scale of folding.
Kim, Namhee; Kang, Jun Su; Jun, Taesuk; Suh, Jong-Min; Roh, Deok-Ho; Park, Won-Woo; Kwon, Oh-Hoon; Kwon, Tae-Hyuk; Ryu, Mi Hee LimDu Yeol; Seo, Myungeun; Kim, Byeong-Su
This study develops a new type of C3-symmetric triphenylene triimide (TTI) bearing different oligo(ethylene glycol) side chains via imide linkages. By exploiting the unique TTI molecule as a building block, supramolecular polymerization is explored based on π–π stacking and hydrophilic/hydrophobic interactions in various solvents and the rates of heating/cooling process. The molecular chirality of the TTI unimer induces a preferential helicity formation in fibrous structures, while the achiral side chain allows the formation of linear nanofibers. The stacking type of supramolecular polymerization is highly dependent on the point chirality of the side chains, as indicated by the spectroscopic analyses, including ultraviolet–visible (UV/vis) and circular dichroism (CD) spectroscopy with atomic force microscopy (AFM), transmission electron microscopy (TEM), and wide-angle X-ray scattering (WAXS). Interestingly, the supramolecular polymerization does not occur in its monomeric state due to the generation of radical anions from the imide groups upon UV irradiation. In contrast, the fibrous structure in the assembled state is maintained, owing to the intermolecular interaction. This study provides a new direction in the phototriggered control of the supramolecular chiral assembly.
Bioadhesives are becoming an essential and important ingredient in medical science. Despite numerous reports, developing adhesive materials that combine strong adhesion, biocompatibility, and biodegradation remains a challenging task. Here, we present a biocompatible yet biodegradable block copolymer-based waterborne superglue that leads to an application of follicle-free hair transplantation. Our design strategy bridges self-assembled, temperature-sensitive block copolymer nanostructures with tannic acid as a sticky and biodegradable polyphenolic compound. The formulation further uniquely offers step-by-step increases in adhesion strength via heating–cooling cycles. Combining the modular design with the thermal treating process enhances the mechanical properties up to 5 orders of magnitude compared to the homopolymer formulation. This study opens a new direction in bioadhesive formulation strategies utilizing block copolymer nanotechnology for systematic and synergistic control of the material’s properties.
Randomness is perceived in two different extremes, in macroscopic homogeneity and local heterogeneity, but apparently far away from order. Here we show that a periodic order spontaneously arises from a binary random copolymer when self-assembly occurs in an ensemble containing > 1015 possible chain sequences. A Bernoullian distribution of hydrophilic and hydrophobic side chains grafted onto a linear backbone was constructed by random copolymerization. When the polymer chains associate in water, a sequence matching problem occurs because of the drastic heterogeneity in sequence: this is believed to generate local curvature mismatches which deviate from the ensemble-averaged interfacial curvature. Periodic folding of the self-assembled bilayer stabilizes the curvature instability as recurring hinges. Reminiscent of chain-folded lamellae found in polymer crystallization, this new liquid crystalline mesophase, characterized as bilayer-folded lamellae, manifests itself as an anisotropically alignable birefringent hydrogel with structural hierarchy across multiple length scales.
Circularly polarized light (CPL) is an inherently chiral entity and is considered one of the possible deterministic signals that led to the evolution of homochirality. While accumulating examples indicate that chirality beyond the molecular level can be induced by CPL, not much is yet known about circumstances where the spin angular momentum of light competes with existing molecular chiral information during the chirality induction and amplification processes. Here we present a light-triggered supramolecular polymerization system where chiral information can both be transmitted and nonlinearly amplified in a “sergeants-and-soldiers” manner. While matching handedness with CPL resulted in further amplification, we determined that opposite handedness could override molecular information at the supramolecular level when the enantiomeric excess was low. The presence of a critical chiral bias suggests a bifurcation point in the homochirality evolution under random external chiral perturbation. Our results also highlight opportunities for the orthogonal control of supramolecular chirality decoupled from molecular chirality preexisting in the system.
Supramolecular polymerization offers a fascinating opportunity to develop dynamic soft materials by associating monomeric building blocks via noncovalent interactions. We report that polymerization can spontaneously drive the supramolecular polymerization of nanoscale micellar objects. We constructed the patchy micelles via two-step polymerization-induced self-assembly. A horizontal association between the patches results in a 1D supermicellar chain in situ by minimizing the enthalpic penalty of exposing the growing chains to solvent. Its length grows with increasing degree of polymerization, confirming that the supramolecular polymerization was triggered and controlled by polymerization. Our results highlight the observation that (1) the entire self-assembly process of forming, compartmentalizing, and associating the micelles can be driven by polymerization in a concerted manner and that (2) polymerization-induced self-assembly now can use compartmentalized nanoobjects as substrates beyond block copolymer chains. Polymerization-induced supramolecular polymerization could be useful for the autonomous preparation of hierarchical nanostructures.
Introduction of asymmetry into a supramolecular system via external chiral stimuli can contribute to the understanding of the intriguing homochirality found in nature. Circularly polarized light (CPL) is regarded as a chiral physical force with right- or left-handedness. It can induce and modulate supramolecular chirality due to preferential interaction with one enantiomer. Herein, this review focuses on the photon-to-matter chirality transfer mechanisms at the supramolecular level. Thus, asymmetric photochemical reactions are reviewed, and the creation of a chiral bias upon CPL irradiation is discussed. Furthermore, the possible mechanisms for the amplification and propagation of the bias into the supramolecular level are outlined based on the nature of the photochromic building block. Representative examples, including azobenzene derivatives, polydiacetylene, bicyclic ketone, polyfluorenes, Cn-symmetric molecules, and inorganic nanomaterials, are presented.
We developed a methodology, inspired by the folding of proteins, for the precision synthesis of hairy polymer nanoparticles. High-molar mass and narrowly dispersed graft copolymers were synthesized by graft-through ring opening metathesis polymerization, to incorporate a designated number of side chains and dimerizable cinnamic acid groups. Intrachain photodimerization collapsed the backbone and arrested it into a compact globular conformation, resulting in hairy nanoparticles topologically equivalent to a core cross-linked star polymer. The single-chain collapse process translates the molecular information written on the 1D graft copolymer into the 3D globular polymer nanoparticle, like protein folding. Unprecedented control over structural parameters was achieved, including the length, number, and composition of the side chains as well as cross-linking density. Different side chains formed distinct subdomains in the sterically congested nanoparticle state and further self-assembled into micellar aggregates in a selective solvent. Both experimental observations and computational simulations indicated that preorganization of the side chains in the block sequence produces subdomains which primarily follow the backbone length scale, while random sequences showed side chain-dependent scaling. Polymer nanoparticles with discrete multiple subdomains were produced by folding of the ternary block graft copolymers. Drastic differences in the self-assembly behavior of ABC- and ACB-sequenced nanoparticles indicate that the spatial organization of subdomains can be achieved by sequence control.
We report the self-assembly of monolayer vesicles from Janus core–shell bottlebrush polymers. A route was developed to synthesize doubly grafted bottlebrush copolymers (DGBCPs) possessing A-b-B and B′-b-C side chains on a single repeating unit. Graft-through ring-opening metathesis polymerization of a norbornene moiety installed by single unit monomer insertion allowed us to place the backbone on any repeating unit of the core (B and B′) block. By decorating each core chain end with different chains via reversible addition–fragmentation chain transfer polymerization, we can obtain nanoobjects with an asymmetric B core and a phase-separated A/C shell. We demonstrate that polystyrene-branch-polystyrene′ and polylactide-b-polystyrene-branch-polystyrene′-b-poly(n-butyl acrylate) macromonomers can be successfully synthesized and polymerized to produce DGBCPs in high yields (81–94% conversion) with an absolute molar mass of 149–395 kg mol–1 and a dispersity of 1.18–1.38. In a solvent slightly more selective to A than C, self-assembly of monolayer vesicles with diameter of <100 nm was observed by transmission electron microscopy. Dissipative particle dynamics simulations suggest that increasing the backbone length and moving the backbone toward the B′/C interface increases the backbone bending energy and favors a lower curvature. The spontaneous curvature appears to prefer a particular layer radius, avoiding bilayer formation.
Heteroarm core cross-linked star (CCS) polymers consist of two different polymer chains covalently joined to a cross-linked core. We investigated their self-assembly behavior to understand whether intramolecular segregation can be induced during synthesis, to produce spatial domains enriched with each polymer, and whether they would exhibit well-defined microphase separation morphologies as a result. Heteroarm CCS polymers containing polylactide (PLA) and polystyrene (PS) arms were synthesized by reversible addition–fragmentation chain transfer copolymerization of styrene and 1,2-bis(maleimidoethane) in the presence of a PLA-macro chain transfer agent (PLA-CTA), followed by chain extension with styrene (the in–out route). Dynamic light scattering, transmission electron microscopy, and small angle X-ray scattering analyses were employed to examine the self-assembly behavior in toluene and acetonitrile, as a relatively neutral and a PLA-selective solvent, respectively. Above a critical PS molar mass, lamellar-like and spherical morphologies were observed, formed by microphase separation into discrete PLA and PS domains. The increase in order with increasing PS molar mass was consistent with the segregation strength-dependent microphase separation behavior. In contrast, when the CCS polymer was synthesized by simultaneously joining PLA and PS chains (the multi macroinitiatior route) it produced rather ill-defined self-assemblies, suggesting that styrene chain extension via the in–out process is important to achieve intramolecular segregation. Using the more PLA-selective acetonitrile as a polymerization solvent indeed produced more well-defined supermicelles with PS cores and PLA coronas, confirming that intramolecular segregation can be driven by the incompatibility of the growing PS to the intramolecular environment, including PLA and the solvent.
Partially sulfonated amphiphilic poly(arylene ether sulfone)s (PSPAESs) were synthesized by one-step nucleophilic aromatic substitution copolymerization. A 4-fluoro-4′-hydroxydiphenyl sulfone potassium salt was used as a hydrophobic monomer, and 5-((4-fluorophenyl)sulfonyl)-2-hydroxybenzenesulfonic acid as a hydrophilic monomer bearing a sulfonic acid group was synthesized from the hydrophobic monomer via selective sulfonation. 1H and 13C nuclear magnetic resonance spectroscopy analysis of PSPAESs indicated formation of statistical amphiphilic copolymers with control over the degree of sulfonation by varying the feed. Dynamic light scattering and transmission electron microscopy analysis indicated that PSPAESs self-assembled into spherical micelles in aqueous solutions. Interestingly, the micellar solution of PSPAESs prepared by dialysis was found to grow Cu2S nanowires on a Cu grid under ambient conditions. Formation of Cu2S nanowires on various substrates including a Si wafer and graphene was demonstrated in the presence of Cu and a sulfur source. UV-vis spectroscopy and X-ray photoelectron spectroscopy data suggests PSPAESs assist dissolution of metallic Cu into Cu(II) enabling the formation of Cu2S nanowires.
Evolution of supramolecular chirality from self-assembly of achiral compounds and control over its handedness is closely related to the evolution of life and development of supramolecular materials with desired handedness. Here we report a system where the entire process of induction, control and locking of supramolecular chirality can be manipulated by light. Combination of triphenylamine and diacetylene moieties in the molecular structure allows photoinduced self-assembly of the molecule into helical aggregates in a chlorinated solvent by visible light and covalent fixation of the aggregate via photopolymerization by ultraviolet light, respectively. By using visible circularly polarized light, the supramolecular chirality of the resulting aggregates is selectively and reversibly controlled by its rotational direction, and the desired supramolecular chirality can be arrested by irradiation with ultraviolet circularly polarized light. This methodology opens a route to ward the formation of supramolecular chiral conducting nanostructures from the self-assembly of achiral molecules.