Multicompartment polymer nanoparticles, such as two-faced Janus and patchy particles composed of distinct chemical features, have received increasing attention because of their utility for interfacial and self-assembly applications originating from the asymmetric particulate structure. This review discusses such nanoparticles in several tens of nanometers produced by controlled polymerizations, which enable scalable synthesis with control of molecular characteristics. We focus on miktoarm core cross-linked star polymers and Janus core–shell bottlebrush polymers as 0D and 1D anisotropic nano-objects containing a discrete core and a compartmentalized shell. We discuss how controlled polymerizations can covalently build such complex architectures with spatial control of the constituting segments to achieve intramolecular segregation. Then, we collectively view their distinct interfacial and self-assembling behaviors reported in the literature from experimental and simulation perspectives.
Macromolecular self-assembly is essential in life and interfacial science. A macromolecule consisting of chemically distinct components tends to self-assemble in a selective solvent to minimize the exposure of the solvophobic segments to the medium while the solvophilic segments adopt extended conformations. While micelles composed of linear block copolymers represent classic examples of such solution assembly, recent interest focuses on the self-assembly of complex macromolecules with nonlinear architectures, such as star, graft, and bottlebrush. Such macromolecules include several to hundreds of polymer chains covalently tied to a core and a backbone. The pre-programmed, non-exchangeable chain arrangement makes a huge difference in their self-assembly. The field has witnessed tremendous advances in synthetic methodologies to construct the desired architectures, leading to discoveries of exotic self-assembly behavior. Thanks to the rapid evolution of computing power, computer simulation has also been an emerging and complementary approach for understanding the association mechanism and further predicting the self-assembling morphologies. However, simulating the self-assembly of architected macromolecules has posed a challenge as a huge number of objects should be included in the simulations. Comparing experimental results with simulations is not always straightforward, as synthetic routes to well-defined model systems with systematically controlled structural parameters are not often available. In this manuscript, we propose to bridge a gap between experiments and simulations in self-assembly of architected macromolecules. We focus on the key articles in this area reporting experimental evidence and simulation details and also cover recent examples in the literature. We start with discussing simulation methodologies applicable to investigate solution self-assembly across multiple levels of chemical resolution from all-atom to particle dynamics. Then, we delve into topological design, synthesis, and simulation of nonlinear macromolecules, including dendritic/star, network, and graft/bottlebrush polymers, to understand the architectural effect on the self-assembly behavior. We expand our discourse to embrace recent advances toward realizing more complex systems. For example, self-assembly in the presence of strong Coulombic interactions, such as in the case of polyelectrolytes, geometric constraints, and other components in solutions, exemplified by inorganic fillers, are introduced. Finally, the challenges and perspectives are discussed in the final section of the manuscript.
We report a synthetic methodology for decorating a surface of metal–organic frameworks (MOFs) with polymers through postsynthetic modification. Well-defined polymers with reversibly deactivated radical species at their chain end were reacted with vinyl-functionalized MOFs in the presence of a radical initiator. The radical addition forms a C–C bond between the polymer end with the functional group at the MOF ligand. We used sterically bulky star polymers containing electron-deficient maleimide chain ends, which facilitated modification of the external surface, yielding polymer-grafted MOF composite particles. A patchy MOF particle can also be obtained by simultaneously grafting two polymers and jammed at the immiscible liquid–liquid interface. We further show that the selective removal of a sacrificial polymer would partially expose the surface of MOFs to external environment, which hinders the uptake of macromolecular guests above the critical hydrodynamic size. Overall, four polymer@MOF composites have successfully been achieved through the present postsynthetic patchworks on MOFs with star polymers and selective etching process.
Supramolecular polymerization offers a fascinating opportunity to develop dynamic soft materials by associating monomeric building blocks via noncovalent interactions. We report that polymerization can spontaneously drive the supramolecular polymerization of nanoscale micellar objects. We constructed the patchy micelles via two-step polymerization-induced self-assembly. A horizontal association between the patches results in a 1D supermicellar chain in situ by minimizing the enthalpic penalty of exposing the growing chains to solvent. Its length grows with increasing degree of polymerization, confirming that the supramolecular polymerization was triggered and controlled by polymerization. Our results highlight the observation that (1) the entire self-assembly process of forming, compartmentalizing, and associating the micelles can be driven by polymerization in a concerted manner and that (2) polymerization-induced self-assembly now can use compartmentalized nanoobjects as substrates beyond block copolymer chains. Polymerization-induced supramolecular polymerization could be useful for the autonomous preparation of hierarchical nanostructures.
We developed a methodology, inspired by the folding of proteins, for the precision synthesis of hairy polymer nanoparticles. High-molar mass and narrowly dispersed graft copolymers were synthesized by graft-through ring opening metathesis polymerization, to incorporate a designated number of side chains and dimerizable cinnamic acid groups. Intrachain photodimerization collapsed the backbone and arrested it into a compact globular conformation, resulting in hairy nanoparticles topologically equivalent to a core cross-linked star polymer. The single-chain collapse process translates the molecular information written on the 1D graft copolymer into the 3D globular polymer nanoparticle, like protein folding. Unprecedented control over structural parameters was achieved, including the length, number, and composition of the side chains as well as cross-linking density. Different side chains formed distinct subdomains in the sterically congested nanoparticle state and further self-assembled into micellar aggregates in a selective solvent. Both experimental observations and computational simulations indicated that preorganization of the side chains in the block sequence produces subdomains which primarily follow the backbone length scale, while random sequences showed side chain-dependent scaling. Polymer nanoparticles with discrete multiple subdomains were produced by folding of the ternary block graft copolymers. Drastic differences in the self-assembly behavior of ABC- and ACB-sequenced nanoparticles indicate that the spatial organization of subdomains can be achieved by sequence control.
This study explores hyper-cross-linking of the cores of block copolymer micelles as a means to generate star polymers with a hyper-cross-linked core surrounded by linear corona arms. A solution of poly(methyl methacrylate)-b-polystyrene (MS) was prepared in acetonitrile to form micelles which were reacted with α,α’-dichloro-p-xylene in the presence of FeCl3 to produce core hyper-cross-linked star (CHS) polymers by selective cross-linking of the PS core. A kinetic investigation showed formation of high-molar mass species (>104 kg mol−1) within 1 h of reaction, which supported conversion of individual MS micelles into CHS polymers. We synthesized several CHS polymers by varying the PS core fractions from 20 to 53%. All the polymers possessed discrete spherical cores that were 19–60 nm in diameter and all were highly soluble in organic solvents retaining the CHS architecture. While permanent microporosity was not detected by gas sorption measurements, increased dye uptake of CHS polymer in solution suggests utility of CHS polymers as stable and solution-processible nanocontainers with accessible free volume in the core.
We report polymerization-induced self-assembly via controlled cross-linking copolymerization to produce robust block copolymer micelles with spherical, elongated, and branched shapes. Reversible addition–fragmentation chain transfer (RAFT) copolymerization of styrene and divinylbenzene (DVB) or 1,2-bismaleimidoethane (BMI) as a cross-linker in the presence of a polylactide macro-chain transfer agent (PLA-CTA) was performed in acetonitrile, which is a non-solvent to polystyrene (PS). The addition of the cross-linker accelerates the copolymerization compared to styrene homopolymerization, which leads to the formation of block polymer micelles within a shorter time frame, followed by in situ inter-chain cross-linking. The micelles are virtually identical to the core cross-linked star polymer consisting of a cross-linked polystyrenic core surrounded by a PLA corona. Molecular weights up to more than 1000 kg mol−1 could be obtained with relatively narrow dispersity values (1.1–1.4). In the case of copolymerization with DVB, the micellar morphology changes from spherical to elongated and branched shapes with increasing conversion. The size and morphology of the micelles are retained in a good solvent to PS, suggesting that the in situ cross-linking effectively stabilizes the micellar core. BMI undergoes alternating copolymerization with styrene in the early stage of polymerization and yields spherical micelles exclusively, because the densely cross-linked core seems to prevent further morphological transition.
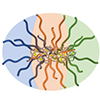
Heteroarm core cross-linked star (CCS) polymers consist of two different polymer chains covalently joined to a cross-linked core. We investigated their self-assembly behavior to understand whether intramolecular segregation can be induced during synthesis, to produce spatial domains enriched with each polymer, and whether they would exhibit well-defined microphase separation morphologies as a result. Heteroarm CCS polymers containing polylactide (PLA) and polystyrene (PS) arms were synthesized by reversible addition–fragmentation chain transfer copolymerization of styrene and 1,2-bis(maleimidoethane) in the presence of a PLA-macro chain transfer agent (PLA-CTA), followed by chain extension with styrene (the in–out route). Dynamic light scattering, transmission electron microscopy, and small angle X-ray scattering analyses were employed to examine the self-assembly behavior in toluene and acetonitrile, as a relatively neutral and a PLA-selective solvent, respectively. Above a critical PS molar mass, lamellar-like and spherical morphologies were observed, formed by microphase separation into discrete PLA and PS domains. The increase in order with increasing PS molar mass was consistent with the segregation strength-dependent microphase separation behavior. In contrast, when the CCS polymer was synthesized by simultaneously joining PLA and PS chains (the multi macroinitiatior route) it produced rather ill-defined self-assemblies, suggesting that styrene chain extension via the in–out process is important to achieve intramolecular segregation. Using the more PLA-selective acetonitrile as a polymerization solvent indeed produced more well-defined supermicelles with PS cores and PLA coronas, confirming that intramolecular segregation can be driven by the incompatibility of the growing PS to the intramolecular environment, including PLA and the solvent.
We explored reversible addition-fragmentation chain transfer (RAFT) copolymerization of 1,2-bis(maleimidoethane) (BMI) with styrene (S) in the presence of polylactide macro-chain transfer agent (PLA-CTA) as a means to synthesize heteroarm core cross-linked star (CCS) polymers consisting of PLA and PS arms (PLAnPSn). Because of the strong alternating tendency of maleimide and styrenic double bonds, copolymerization of BMI with an excess of S depleted BMI in the early stage of polymerization forming a cross-linked core. The remaining S was successively polymerized to grow PS arms from the core, completing PLAnPSn via “in–out” mechanism. Use of a stoichiometric amount of S produced PLAn, which could be used as a macro-CTA for the synthesis of more well-defined PLAnPSn. Compared with divinylbenzene, copolymerization of BMI with S was much more effective for core formation suggesting the importance of the alternating character of the copolymerization. While PLAnPSn existed as stable nanoparticles in a neutral solvent in contrast to linear PLA-b-PS, it also self-assembled to form microphase-separated structures in a selective solvent and in bulk indicating that PLA and PS arms can be intramolecularly segregated.